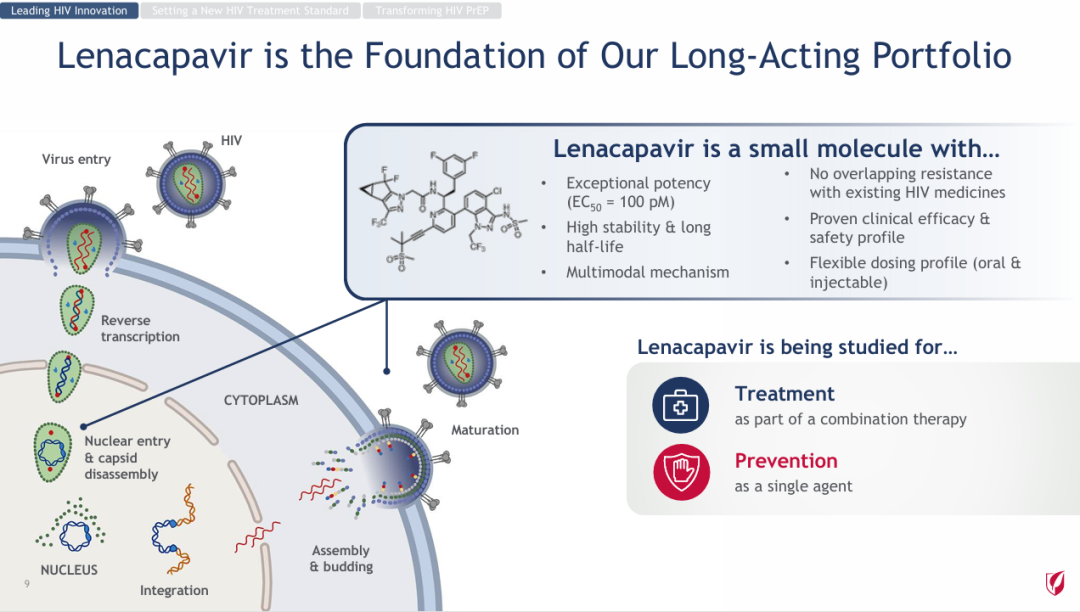
The high risk of clinical development for innovative drugs is an industry consensus, but even so, pharmaceutical companies continue to persevere, investing substantial resources each year into conducting numerous clinical studies. Despite the significant investment, there are also many new drugs reporting successful trial results.
These clinical outcomes have driven continuous breakthroughs in various diseases, constantly renewing the treatment landscape and bringing more precious hope to patients. PharmaCube has specially selected 10 "milestone" clinical studies from 2024 for your reference.
Top 10 Clinical Studies of Milestone Significance in 2024
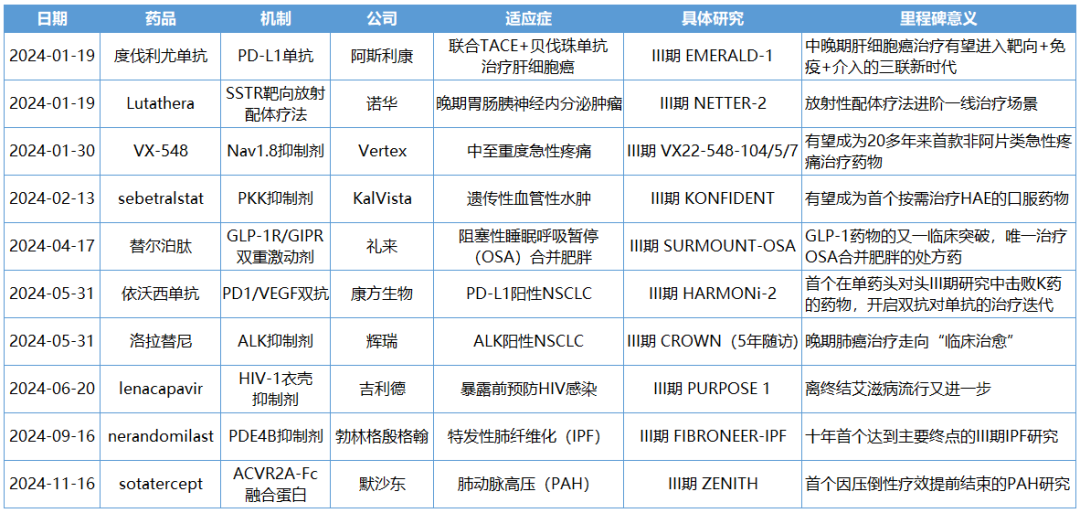
NO.1 Durvalumab EMERALD-1 Study
Company: AstraZeneca
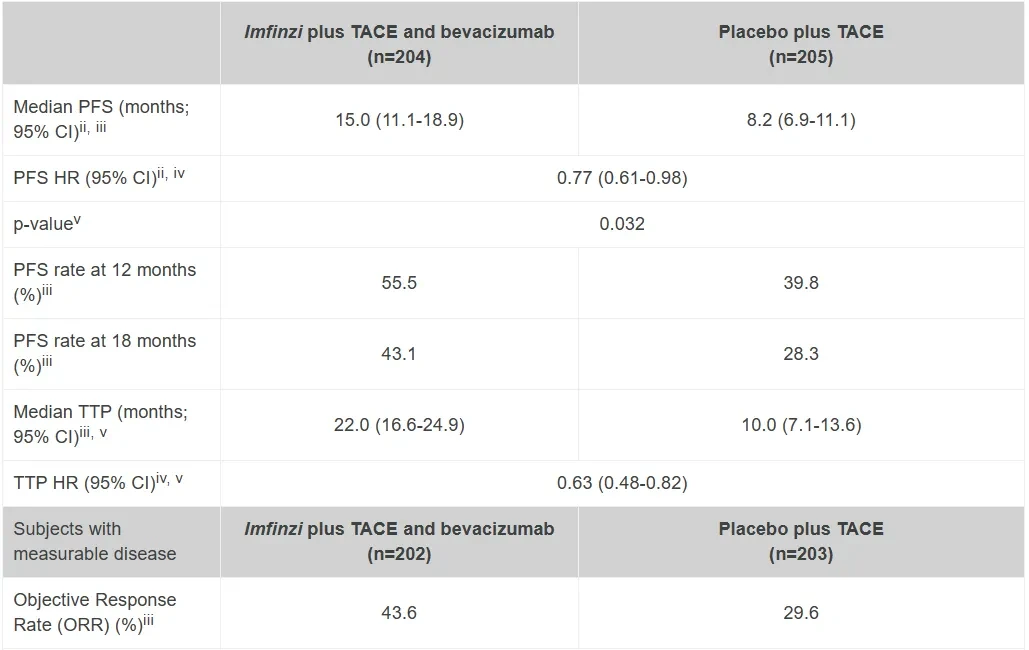
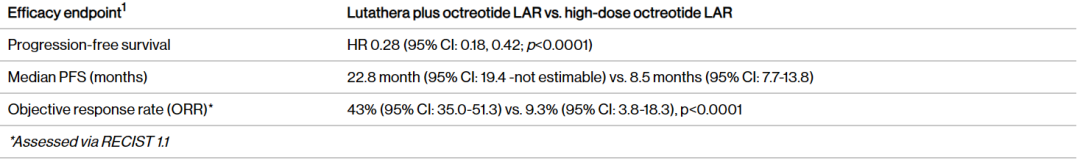
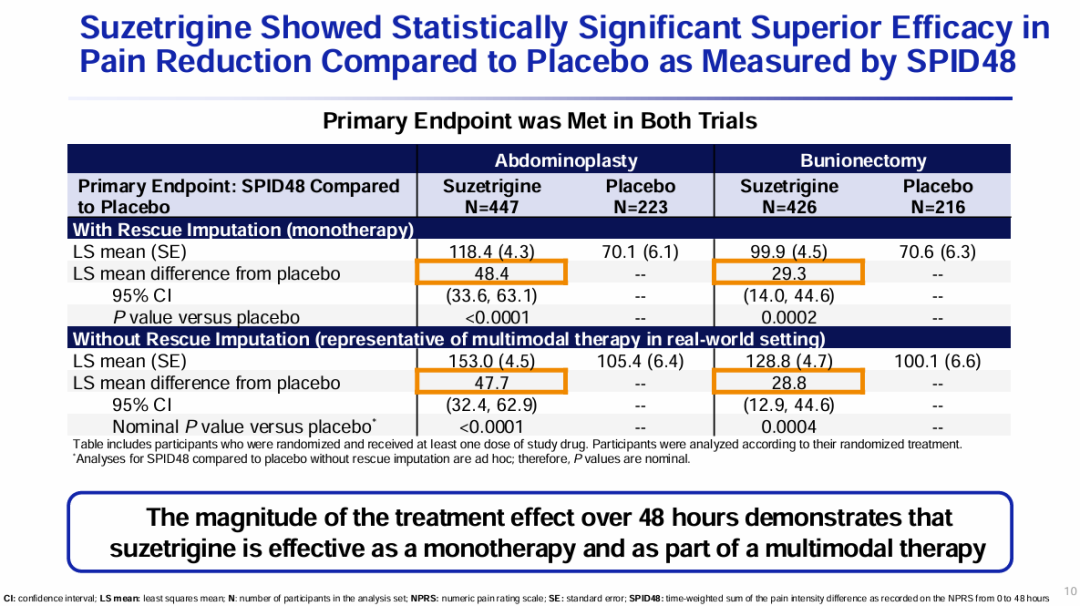
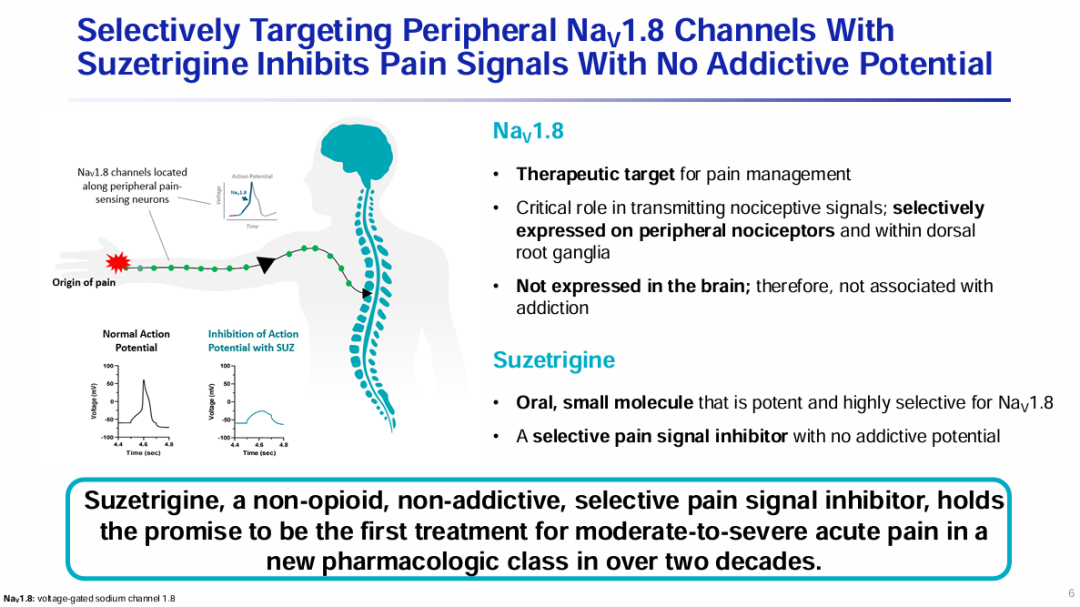
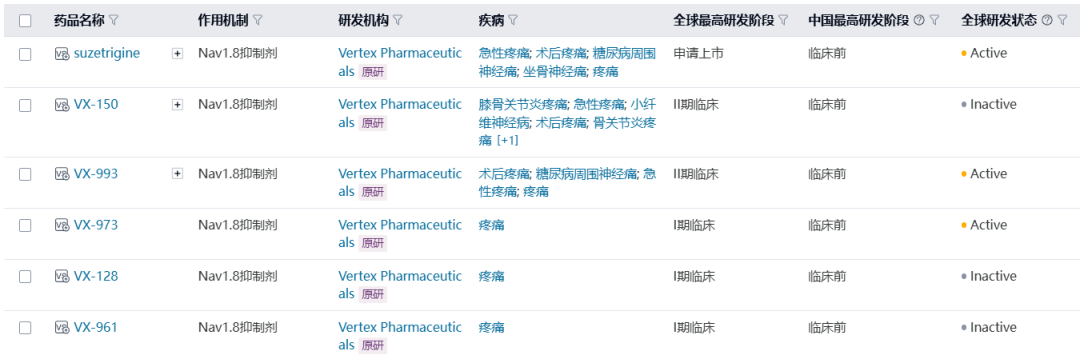
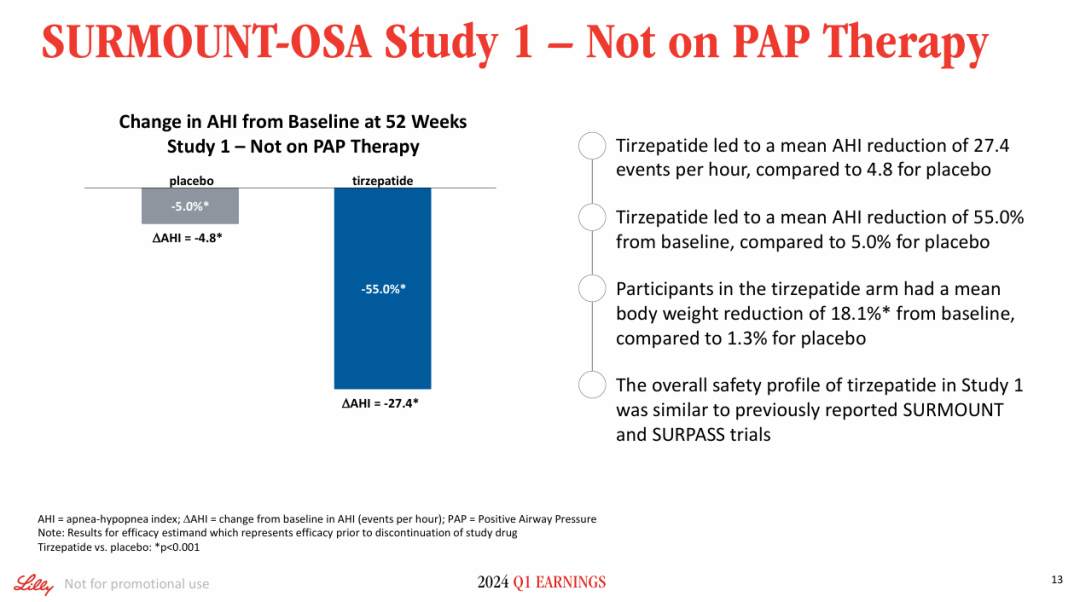
Milestone Significance: Targeted + Immunotherapy + Intervention Triple Therapy Validated for the First Time in Advanced Hepatocellular CarcinomaOn January 19, AstraZeneca announcedDurvalumab (Imfinzi) in combination withTransarterial Chemoembolization (TACE)and BevacizumabTreatmentSuitable for embolizationPatients with Hepatocellular Carcinoma (HCC)Phase III EMERALD-1 Study Yields Positive Results.InPrimary EndpointIn terms of progression-free survival (PFS), compared with TACE alone,DurvalumabCombination Therapy GroupShowed statistically significant and clinically meaningful improvements.DurvalumabThe median PFS in the combination therapy group was 15 months, compared to 8.2 months in the TACE treatment group. The observed PFS benefit was generally consistent across pre-specified key subgroups.Compared with TACE alone,DurvalumabCombination therapy reduced the risk of disease progression or death by 23% (hazard ratio[HR]: 0.77; 95% Confidence Interval [CI] 0.61-0.98; p=0.032).Hepatocellular carcinoma is the most common type of liver cancer, with approximately 20%-30% of hepatocellular carcinoma patients being eligible for transarterial chemoembolization (TACE) treatment. TACE is a procedure that blocks the blood supply to the tumor and can also deliver chemotherapy or radiation therapy directly to the liver. Although TACE is the standard treatment in this context, the majority of patients who undergo TACE treatment experience disease progression or recurrence within 8 months.The EMERALD-1 study is the first global Phase III trial to evaluate TACE combined with targeted immunotherapy for hepatocellular carcinoma, achieving positive results.This means that the treatment of advanced hepatocellular carcinoma is expected to enter a new era of triple therapy: targeted + immunotherapy + interventional therapy.There is no doubt that the triple therapy for hepatocellular carcinoma is a valuable exploration direction, and more companies are making plans. In April this year, Akeso registered.Comparison of CTLA4/PD1 Bispecific Cadonilimab + Lenvatinib + TACE TreatmentPhase III Study of TACE in Hepatocellular Carcinoma (NCT06371157) is Expected to Complete the Evaluation of the Primary Endpoint PFS by October 2025.At the ESMO Congress in September, the Phase III LEAP-012 study of Keytruda (K药) made a striking appearance. The results showed that the combination of Keytruda, Lenvatinib, and TACE achieved a significant PFS benefit compared to TACE alone (14.6 vs 10.0 months).NO.2 LutatheraNETTER-2 StudyMilestone Significance: Radioligand Therapy Joins the Ranks of First-Line TreatmentsOn January 19, Novartis announced its first-in-classSomatostatin Receptor (SSTR)-TargetedData from the Phase III NETTER-2 trial of the radioligand therapy Lutathera shows that, compared to high-dose long-acting release octreotide alone, the combination of Lutathera and long-acting octreotide as first-line treatment canSSTRPatients with advanced grade 2 and 3 well-differentiated gastrointestinal pancreatic neuroendocrine tumors (GEP-NETs) experienced a 72% reduction in the risk of disease progression or death.Lutathera Becomes the First Radioactive Ligand Therapy to Show Clinically Meaningful Benefit in First-Line Treatment, also inThe therapy with the best overall efficacy and safety in the treatment of GEP-NETs.In the Phase III NETTER-2 trial,Lutathera CombinationOctreotide significantly prolonged the median PFS to 22.8 months in newly diagnosed grade 2 and grade 3 advanced GEP-NETs patients, while the median PFS was 8.5 months in the high-dose octreotide monotherapy group.Neuroendocrine tumors (NETs) are cancers that originate from neuroendocrine cells throughout the body and are generally considered to be slow-growing malignancies. However, some NETs progress rapidly with poor prognosis, and in many cases, are not diagnosed until the disease has advanced to a late stage. Although NETs are rare diseases, their incidence has increased by more than 500% over the past 30 years, creating an urgent need for more treatment options for newly diagnosed patients with inoperable or advanced stages.Lutathera was approved by the FDA for marketing in 2018 to treat adult patients with SSTR-positive GEP-NETs and has expanded its target population this year to include children aged 12 years and above. In 2023, Lutathera's total annual sales reached $605 million, with sales in the first three quarters of this year amounting to $534 million.As a leader in the field of nuclear medicine, Novartis has rapidly built up relevant technology platforms by integrating internal and external innovations, creating two blockbuster products, Lutathera and Pluvicto, with more promising pipelines underway.SSTR2-TargetedDevelopment of Radioligand Therapy, Novartis in November this year withRatio Therapeutics has reached an exclusive global cooperation.NO.3 VX-548Three Phase III StudiesMilestone Significance: Expected to Become the First New Mechanism Drug for Acute Pain Treatment in Over 20 YearsJanuary 30,Vertex Pharmaceuticals, Inc. announced its potential first-in-class NaV1.8 inhibitor VX-548 (suzetrigine)Positive Results from Phase III Program for the Treatment of Moderate to Severe Acute PainThis Phase III program includes three Phase III studies (VX22-548-104, VX22-548-105 andVX22-548-107)。VX22-548-104 (NAVIGATE 1, n=1073) and VX22-548-105 (NAVIGATE 2, n=1118) are two randomized, double-blind, placebo-controlled, pivotal clinical trials, with the former targeting patients undergoing bunionectomy and the latter targeting patients undergoing abdominoplasty.This is also the largest Phase III randomized controlled study in the field of acute pain to date.In studies 104 and 105, compared with the placebo group,VX-548 GroupTime-Weighted Sum of Pain Intensity Over 0-48 Hours for the Primary Endpoint(SPID48)Statistically significant improvement in aspects, as evidenced by a clinically meaningful reduction in the Numerical Pain Rating Scale (NPRS) scores from baseline to 48 hours in patients treated with VX-548.Key Secondary Endpoints of the Two Studies – Time to Meaningful Pain Relief (i.e., the time for NPRS scores to decrease by ≥2 points from baseline compared to the placebo group)Successfully Achieved。However, neither Study 104 nor Study 105 metVX-548 is superior to SPID48 in terms of efficacy.Another name for Dihydrocodeine Bitartrate/Acetaminophen (HB/APAP)Key Secondary Endpoint。VX22-548-107 (n=256) isA single-arm clinical trial, the recruited population includesSurgical and non-surgical pain patients.In the 107 study, treatment effectiveness was measured by Patient Global Assessment (PGA), with 83.2% of patients rating VX-548 as good, very good, or excellent for pain relief.VX-548 is a non-opioid, non-addictive selective pain signal inhibitor, with the potential to become the first novel drug for treating moderate to severe acute pain in over 20 years.Vertex believes that NaV1.8, the target of VX-548, is an ideal target for pain treatment. On one hand, NaV1.8 plays a key role in transmitting pain signals and is selectively expressed in peripheral nociceptors and dorsal root ganglia; on the other hand, this target is not expressed in the brain, thus avoiding the risk of addiction. The biggest risk of opioid analgesics is their addictive nature.It is precisely because of the high potential of this target that Vertex has heavily invested in the research and development of NaV1.8 inhibitors, experiencingVX-128、VX-150、VX-961 Multiple TimesTrial and error, but continue to explore.Now, VX-548 has entered the application stage for market approval, with the indication applied for being moderate to severe acute pain. The PDUFA date is January 30, 2025.Notably, VX-548Treatment of Painful Lumbosacral Radiculopathy (LSR)Phase IIThe study has also recently reached its primary endpoint., the efficacy was less than expected, and the difference between the treatment group and the placebo group was not significant enough. However, Vertex still believes that VX-548 can proceed to Phase III.In the LSR study, it believes that "the placebo effect can be better controlled through innovative clinical trial design."In addition to VX-548, Vertex has other assets in this space, with the next-generation NaV1.8 inhibitors VX-993 and VX-973 also entering clinical trials. For VX-993, Vertex plans to develop not only an oral formulation but also an intravenous infusion version.
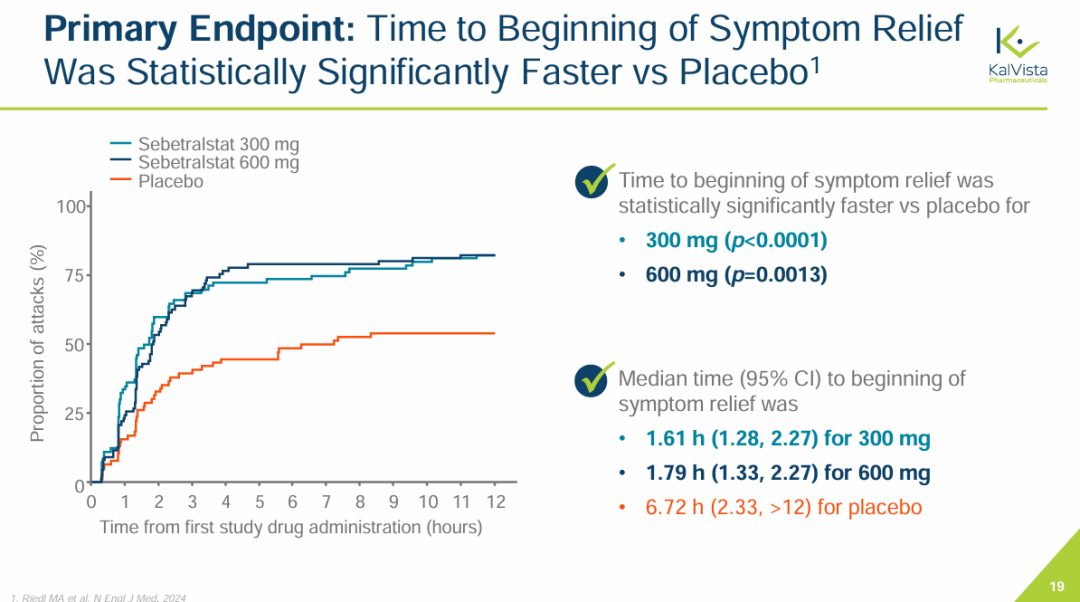
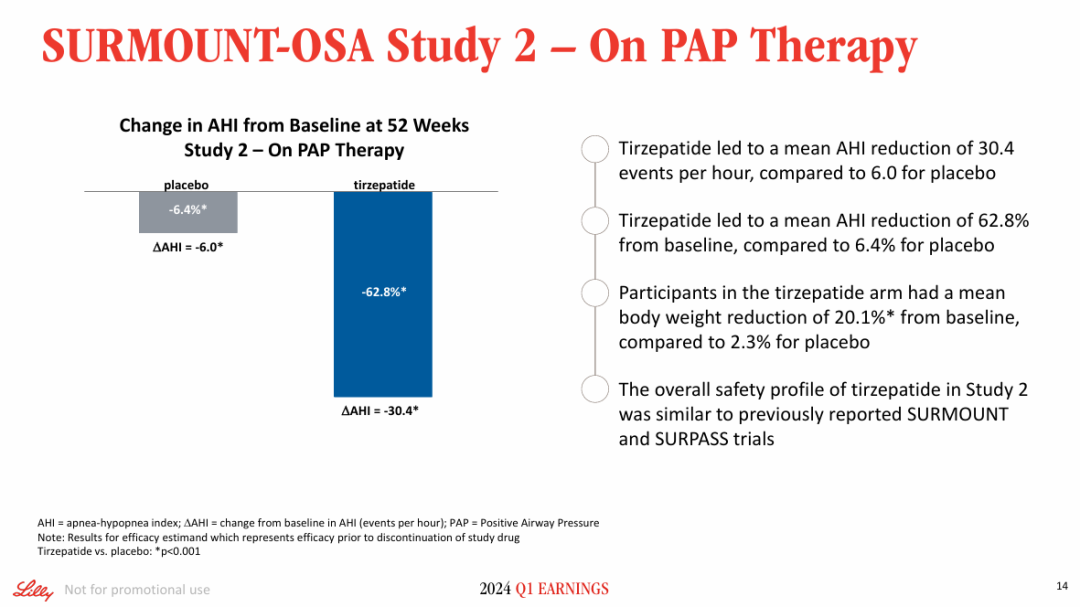
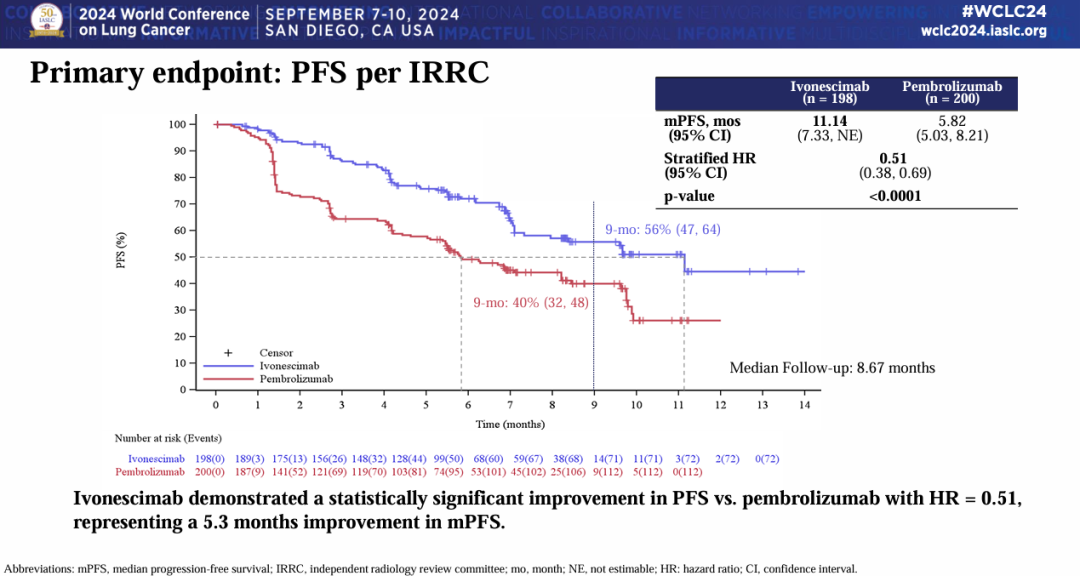
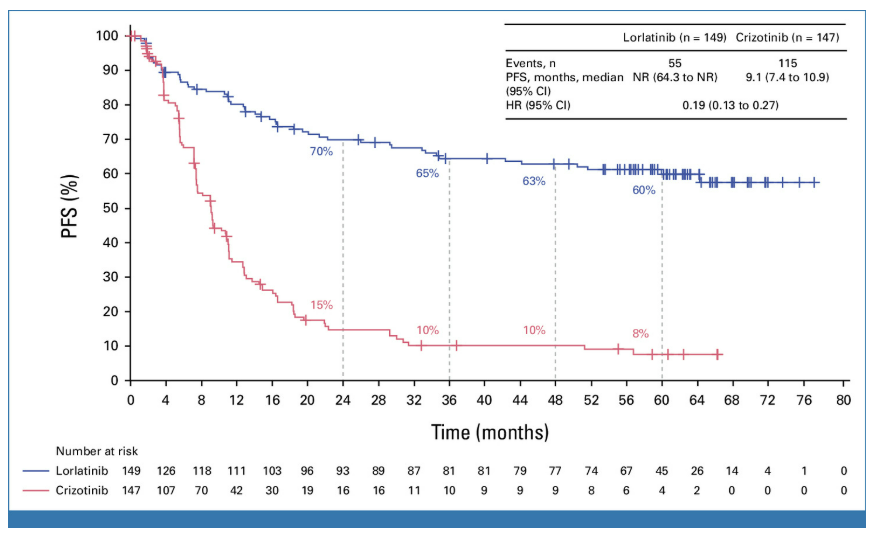
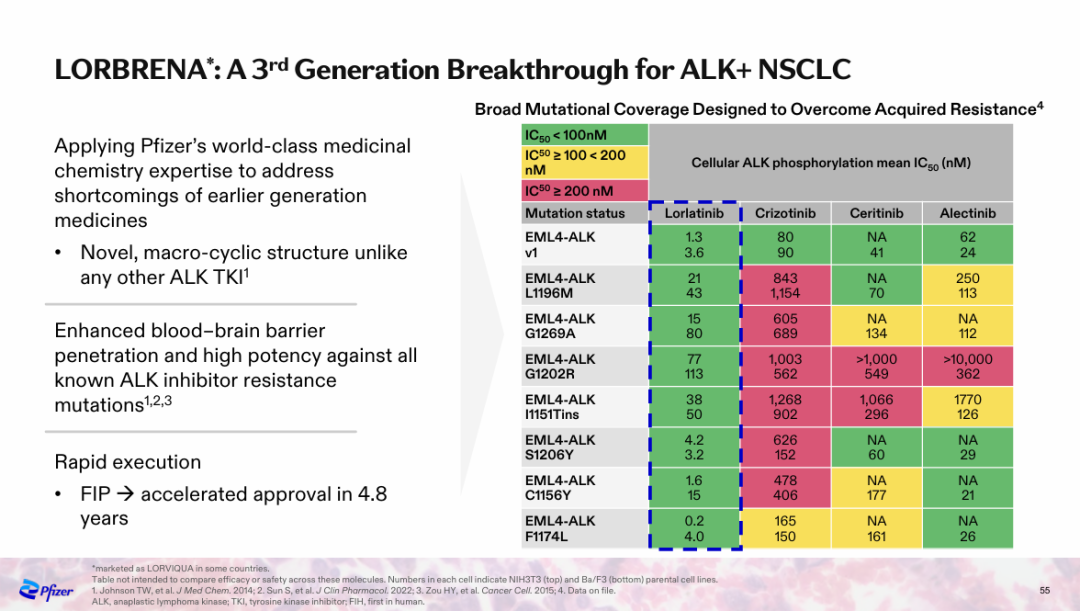
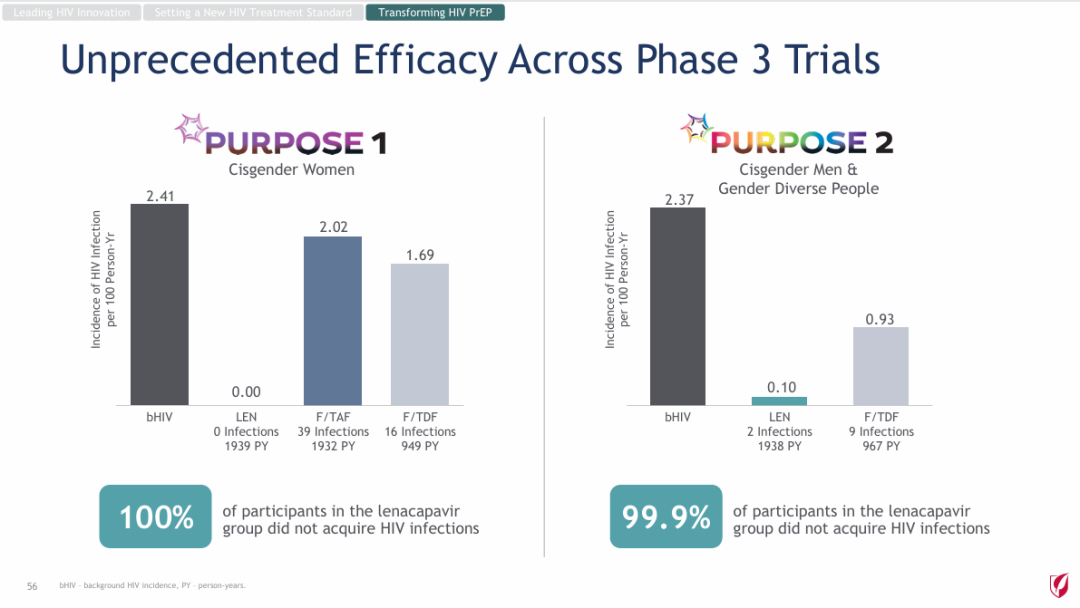
NO.4 sebetralstatKONFIDENT StudyMilestone Significance: Expected to Become the First On-Demand Oral Treatment for HAEOn February 13, KalVista Pharmaceuticals announced positive results from the Phase III KONFIDENT clinical trial, indicating that sebetralstat, an oral plasma kallikrein (PKK) inhibitor for on-demand treatment of hereditary angioedema (HAE), demonstrated significant statistical and clinical efficacy. KONFIDENT is the largest and most representative trial conducted for HAE to date, including adolescents, patients using long-term prophylactic medications, and patients with all attack severities and locations.KONFIDENT Trial Meets All Primary and Key Secondary Endpoints. Treatment with 300mg and 600mg Sebetralstat for HAE Attacks Shows Significant Faster Relief of Symptoms Compared to Placebo. The median time to onset of symptom relief was 1.61 hours for the sebetralstat 300mg group, 1.79 hours for the sebetralstat 600mg group, and 6.72 hours for the placebo group.Consistent with previous studies, sebetralstat was well-tolerated, and its safety profile was similar to placebo. No patients withdrew due to any adverse events, and no serious adverse events (SAEs) related to treatment were observed. The incidence of treatment-related adverse events was 2.3% in the 300mg sebetralstat group, 2.2% in the 600mg sebetralstat group, and 4.8% in the placebo group.Based on the aforementioned research, the FDA has accepted the application for sebetralstat's market approval for the on-demand treatment of HAE attacks in adults and adolescents aged 12 years and above. The PDUFA date is June 17, 2025. If approved,Sebetralstat will become the first on-demand oral medication for HAE.。HAE is a rare genetic disorder that causes a deficiency or dysfunction of the C1 esterase inhibitor (C1INH) protein, followed by uncontrolled activation of the kallikrein-kinin system. Patients with HAE experience painful and debilitating episodes of tissue swelling, which can be life-threatening depending on the affected body parts.HAE Treatment is Divided into Three Categories: On-Demand Treatment for Acute Edema Attacks, Long-Term Prophylaxis (LTP), and Short-Term Prophylaxis (STP). Currently, all approved on-demand treatment regimens require either intravenous or subcutaneous administration. Therefore, a safe and effective oral on-demand treatment would be highly valuable in addressing unmet needs and reducing the treatment burden associated with injectable therapies.NO.5 TirzepatideSURMOUNT-OSA StudyMilestone Significance: Marks Another Clinical Breakthrough for GLP-1 Drugs, and the Only Prescription Medication for Treating OSA Combined with ObesityOn April 17, Eli Lilly announced itsGLP-1R/GIPR Dual AgonistTirzepatide (Tirzepatide,Zepbound)For the treatment of moderate to severeObstructive Sleep ApneaPositive Results Obtained from Phase III SURMOUNT-OSA Clinical Trial of OSA Combined with Obesity.OSA is a sleep-related breathing disorder characterized by complete or partial collapse of the upper airway during sleep, leading to apnea or hypopnea, and potentially causing reduced blood oxygen saturation and/or arousals from sleep. OSA can lead to severe cardiometabolic complications, including hypertension, coronary heart disease, stroke, heart failure, atrial fibrillation, and type 2 diabetes.Compared with placebo,TirzepatideSignificantly reduced the Apnea-Hypopnea Index (AHI) in subjects, achieving the primary endpoint. AHI records the number of times per hour of sleep that a subject experiences restricted breathing or complete airflow blockage, and is used to evaluate the severity of OSA and the effectiveness of treatment outcomes.SURMOUNT-OSA Study 1 evaluatedTirzepatideWithout receivingPositive Pressure VentilationEfficacy of (PAP) treatment in adult patients with moderate to severe OSA and obesity. The results showed that at 52 weeks,TirzepatideGroup of SubjectsThe mean AHI decreased by 27.4 events/hour from baseline, compared to a decrease of 4.8 in the placebo group.Times/hour。SURMOUNT-OSA Study 2 evaluatedTirzepatideEfficacy in adult patients with moderate to severe OSA and obesity who accepted and planned to continue PAP therapy. The results showed that at 52 weeks,TirzepatideThe average AHI in the treatment group decreased by 30.4 events/hour from baseline, while the placebo group showed an average decrease of 6.0.Times/Hour。Based on this, the FDA approved tirzepatide in December this year as the first andThe only prescription drug for treating OSA combined with obesity in adult patients。This approvalMeans GLP-1 drugsOpened up another market beyond blood sugar reduction and weight loss.NO.6 IvokisumabHARMONi-2 StudyMilestone Significance: The First Drug to Defeat K-Medicine in a Single-Agent Head-to-Head Phase III Study, Establishing a Clear New Direction for PD-1 Drug UpgradesOn May 31, Akeso announced that the registrational Phase III HARMONi-2 study (AK112-303) evaluating the first-in-class PD-1/VEGF bispecific antibody, ivonescimab monotherapy versus pembrolizumab as a first-line treatment for locally advanced or metastatic non-small cell lung cancer (NSCLC) with positive PD-L1 expression (PD-L1 TPS≥1%), met its primary endpoint of PFS.HARMONi-2 is the world's first Phase III clinical study in which a monotherapy demonstrated significantly positive results compared to pembrolizumab monotherapy. Pembrolizumab had already claimed the top position as the global "best-selling drug" in 2023 with $25 billion in sales. The study enrolled a total of 398 participants, among whom 57.8% had PD-L1 TPS scores between 1% and 49%, and 42.2% had PD-L1 TPS scores of 50% or higher.At the WCLC conference in September, Akeso further disclosed data from the study. In the intent-to-treat (ITT) population, the mPFS for the Ivonescimab group and the Pembrolizumab group were 11.14 months and 5.82 months, respectively (HR=0.51, P<0.0001), with the Ivonescimab treatment group showing a 49% reduction in the risk of disease progression/death.Compared with Pembrolizumab, Ivonescimab significantly improved the ORR (50.0% vs 38.5%) and DCR (89.9% vs 70.5%) in first-line treatment for PD-L1 positive NSCLC patients. However, the OS data from this study is not yet mature. If Ivonescimab can also demonstrate a survival benefit, the treatment paradigm will shift comprehensively from monotherapy to bispecific antibodies.Many overseas companies have made early arrangements, which also gives Chinese-produced PD-1/VEGF bispecific antibodies the opportunity to step onto the global stage. Following the success of the HARMONi-2 study,Instil has introducedIMM2510 from Yiming Angke,MSD, with Pembrolizumab in hand, seems to have a sense of crisis.Secured for over $3.2 billionLM-299 by Lixun Pharmaceuticals,BioNTech AcquisitionPrometheus may also have the factor of PM8002 asset enhancement.NO.7 Lorlatinib CROWNResearchMilestone Significance: Advanced ALK-Positive Lung Cancer Treatment Moving Towards "Clinical Cure"On May 31, at the ASCO meeting, Pfizer announced the long-term follow-up results of the Phase III CROWN study in an oral report. This study aims to evaluate the efficacy of the third-generation ALK inhibitor lorlatinib compared with the first-generation ALK inhibitor crizotinib in previously untreated patients with ALK-positive advanced NSCLC.After a median follow-up of five years, the median PFS in the lorlatinib treatment group has not yet been reached according to the investigator's assessment. This represents the longest PFS ever achieved in the field of ALK-positive NSCLC and also sets a new record for the longest PFS with single-agent targeted therapy in advanced lung cancer.Compared with crizotinib, lorlatinib reduced the risk of disease progression or death by 81% in patients.The 5-year PFS rate in the lorlatinib group was 60%, meaning that 60% of patients had not experienced disease progression or death after five years, compared to 8% in the crizotinib group. "No disease progression or death after five years" also implies that these advanced patients have reached "clinical cure."For patients with brain metastases, the efficacy of lorlatinib is further highlighted. This updated analysis shows that lorlatinib reduced the risk of intracranial disease progression by 94%. The median time to intracranial progression has not been reached in the lorlatinib group, while this figure was 16.4 months in the crizotinib group.The remarkable therapeutic effects achieved by Lorlatinib confirm the correctness of Pfizer's molecular design strategy. Pfizer designed Lorlatinib with a unique macrocyclic structure, which offers improved lipophilicity and deep target binding, thereby enhancing blood-brain barrier penetration and increasing efficacy against resistance mutations to other ALK inhibitors.NO.8 lenacapavirPURPOSE 1 StudyMilestone Significance: One Step Closer to Ending the HIV EpidemicOn June 20, Gilead Sciences announced the interim analysis results of the pivotal Phase III PURPOSE 1 trial, indicating that its HIV-1 capsid inhibitor lenacapavir, which requires only two injections per year, demonstrated 100% efficacy in pre-exposure prophylaxis (PrEP) for preventing HIV infection in women.Lenacapavir is a first-in-class long-acting HIV capsid inhibitor that inhibits the replication of HIV-1 by interfering with multiple critical steps in the virus lifecycle. These include the inhibition of viral capsid-mediated uptake, assembly, and release of HIV-1 proviral DNA, as well as the formation of the viral capsid core. Notably, it has no known cross-resistance with other existing drug classes. In 2022, the drug received FDA approval for marketing to treat HIV-1 infection.PURPOSE 1: The study met the key efficacy endpoint, demonstrating that lenacapavir administered once every six months provided superior prevention efficacy compared to once-daily Truvada (emtricitabine 200mg and tenofovir disoproxil fumarate 300mg) and the background incidence of HIV.In the lenacapavir group, there were 0 HIV infection events among 2,134 women (incidence rate: 0.00/100 person-years). In the Truvada group, there were 16 events among 1,068 women (incidence rate: 1.69/100 person-years).This is the first set of data generated from Gilead Sciences' milestone PURPOSE project. The PURPOSE project comprises five HIV prevention trials conducted worldwide and represents the most comprehensive and diverse HIV prevention trial program to date.Another key trial of the project, PURPOSE 2, yielded results this September. The trial aimed to evaluate the PrEP efficacy of lenacapavir among globally diverse populations, including men, transgender men, and transgender women.Compared with the background HIV incidence rate, lenacapavir reduced the HIV infection rate by 96%. Among the 2,180 participants in the lenacapavir group, there were 2 cases of infection, which means 99.9% of the participants did not contract HIV; lenacapavir also showed better treatment effects than Truvada.The significant results achieved by Lenacapavir in these two Phase III studies demonstrate its potential to transform HIV prevention and help end the HIV epidemic. Based on this, Gilead Sciences has stated that it will initiate a series of global regulatory submissions for the drug's use as PrEP before the end of this year.If approved, lenacapavir could provide a crucial new option for HIV prevention, further driving down HIV prevalence and making the goal of ending the HIV epidemic more attainable. While traditional HIV prevention methods are highly effective when taken as prescribed, this drug may help address potential stigma some individuals face when taking oral PrEP pills and improve adherence and persistence in medication use.NO.9 nerandomilastFIBRONEER-IPF StudyCompany: Boehringer IngelheimMilestone Significance: The First to Reach the Primary Endpoint in a DecadePhase III IPF StudySeptember 16,Boehringer Ingelheim announces its potential first-in-classPhase III Trial of PDE4B Inhibitor Nerandomilast for the Treatment of Idiopathic Pulmonary Fibrosis (IPF)FIBRONEER-IPF Study Meets Primary Endpoint, Showing Improvement in Forced Vital Capacity (FVC) at Week 52 Compared to PlaceboIndicators for Measuring Lung Function) Absolute change from baseline.Over the past decade, this is the first Phase III trial to reach its primary endpoint.IPFTest。FIBRONEER-IPF StudyInConducted in more than 30 countries worldwide, enrolling 1,177 patients, this is the largest clinical trial in the IPF field.Based on the results of this study, Boehringer Ingelheim plans to submit a new drug application for nerandomilast to the FDA and other regulatory authorities worldwide for the treatment of IPF, which is expected to further address the urgent needs in this disease area.Idiopathic Pulmonary Fibrosis (IPF) is one of the most common progressive fibrosing interstitial lung diseases, with clinical symptoms including activity-induced shortness of breath, persistent dry cough, and chest discomfort. Although considered a "rare" disease, IPF affects approximately three million people worldwide. The prognosis for this condition is poor, with a median survival of only 3-5 years after diagnosis.However, the clinical treatment options are limited. According to the PharmaCube database, only two innovative drugs, pirfenidone and nintedanib, have been approved globally for treatment.IPF, andThe developer of Nintedanib is Boehringer Ingelheim.NO.10 sotaterceptZENITH StudyMilestone Significance: The First PAH Study Ended Early Due to Overwhelming EfficacyOn November 25, MSD announced that itsFirst CreationACVR2A-Fc Fusion ProteinThe Phase III ZENITH study of sotatercept (Winrevair) for the treatment of adults with pulmonary arterial hypertension (PAH) functional class III or IV at high risk of death met its primary endpoint, which was the time to the first event of morbidity or death (all-cause mortality, lung transplantation, or hospitalization due to PAH worsening for ≥24 hours).In the ZENITH study, sotatercept significantly reduced the risk of morbidity or death events compared to placebo, with statistical significance and clinical relevance. Both treatments were administered on a background of PAH therapy.Based on the strength of these results, the independent data monitoring committee recommended early termination of the ZENITH study and providing all participants with the opportunity to receive sotatercept treatment through the SOTERIA open-label extension study.This is the first study in the PAH field to be terminated early due to overwhelming efficacy demonstrated in an interim analysis.。PAH is a serious progressive disease with high incidence and mortality.,The clinical manifestations areProliferative Remodeling of Small Pulmonary Arteries and Gradual Luminal NarrowingIt is estimated that there were approximately 40 million PAH patients globally in 2021, with a 5-year mortality rate of about 43%.Currently, existing PAH therapies on the market can alleviate patients' conditions by promoting pulmonary vasodilation but are unable to fundamentally address the issue of pulmonary vascular remodeling.Studies show that the imbalance in cell proliferation and anti-apoptosis signaling pathways mediated by members of the transforming growth factor β (TGF-β) superfamily is one of the key mechanisms driving pulmonary vascular remodeling in PAH patients. Activin receptor type 2A (ACVR2A) is one of the members of the TGF-β superfamily. Therefore, drugs targeting ACVR2A represent a potentially effective approach to reversing pulmonary vascular remodeling.Sotatercept is a first-in-class ACVR2A-Fc fusion protein, composed of the extracellular domain of human Activin receptor IIA fused with the Fc domain of IgG1, which can bind and capture TGF-β family ligands.In September 2021, MSD acquired Acceleron Pharma for $11.5 billion, obtaining sotatercept.In March this year, based on the results of the Phase III STELLAR study, sotatercept was approved for marketing in the United States. Since its launch, sotatercept has shown strong growth, with global sales reaching $149 million in about half a year. The rapid increase in sales also reflects the clinical transformative significance of sotatercept in the treatment of PAH.Copyright © 2024 PHARMCUBE. All Rights Reserved.
Welcome to forward, share, and reasonably cite. When citing, please clearly indicate the source of the article in a prominent position.For reprinting, please leave a message to the WeChat Official Account backend or send a message, and indicate the name and ID of the official account.
Disclaimer: The information in this WeChat article is for general reference only and should not be used directly as decision-making content. PharmaCube assumes no responsibility for any loss incurred by any party due to the use of the content herein.