AI Model Ensemble Strategy by Zelixir Significantly Enhances Protein-Peptide Complex Prediction Accuracy

Zelixir
Protein Structure Prediction and Design Service Platform Provider

Dong-E E-Jiao
Dong-E E-Jiao and its series product manufacturer
In the wave of artificial intelligence empowering life sciences, structural biology has ushered in unprecedented breakthroughs. Recently, byZelixir and Dong-E-E-Jiao Co., Ltd.A jointly conducted study,In-depth evaluation of the performance of the new generation AlphaFold3 (AF3) and its derivative models in protein-short peptide complex modelingThe research team systematically analyzed cutting-edge algorithms such as AF3, Protenix, Chai-1, and Boltz-1, and proposed a more accurate prediction strategy forDrug Design, Signal Transduction Research, and Biomolecular EngineeringProvided strong tool support.

So, can AF3 and its next-generation model solve these problems?The team compared the predictive capabilities of five models and explored how to further improve accuracy through a "combination strategy."Let's take a look at their findings together!
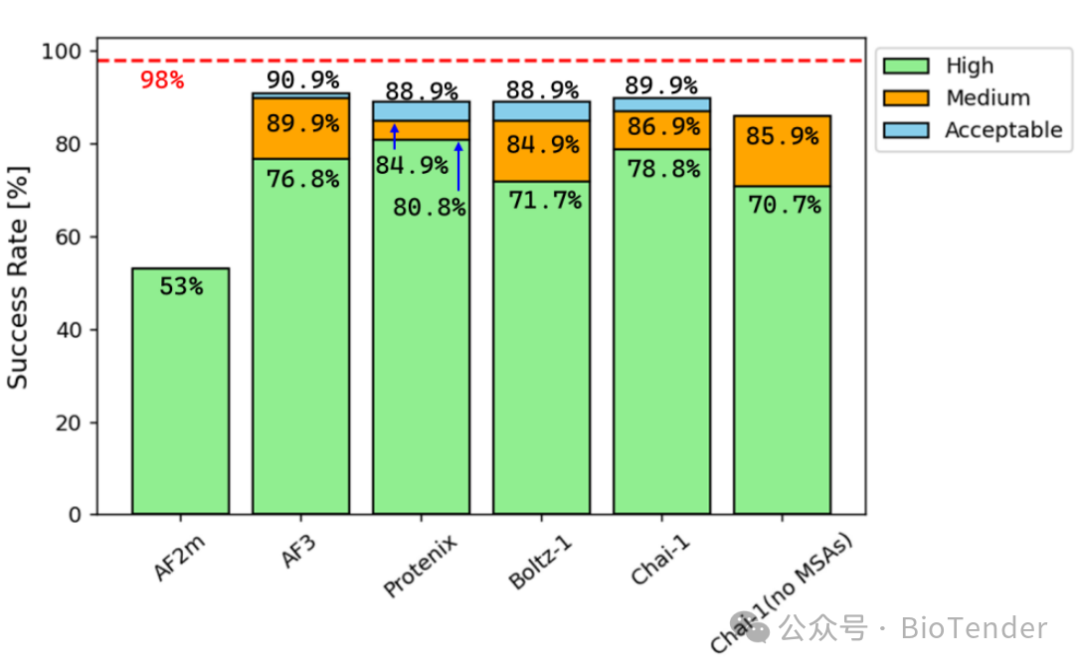
Comparison of success rates across different modeling methods.
Who Will Prevail?
The main objective of this study is:
Evaluate the performance of AF3 and its derivative methods (Protenix, Chai-1, Boltz-1) in protein-short peptide complex prediction and compare them with AF2m. Study the advantages of different models and explore how to improve prediction success rates. Propose an optimized model combination strategy to improve the accuracy of protein-peptide interaction prediction.
In order to scientifically evaluate these models, the research team selected99 Protein-Peptide Complex StructuresAs the benchmark dataset, and precisely measured using multiple key metrics, including:
Fnat (Native Contact Fraction): Measures the similarity between the predicted result and the true crystal structure. Fnat ≥ 0.8 indicates high-precision prediction.
DockQ: Comprehensive evaluation of the quality of protein-protein or protein-short peptide docking models.
Scores such as pLDDT and ipTM are used to assess the reliability of predictive models.
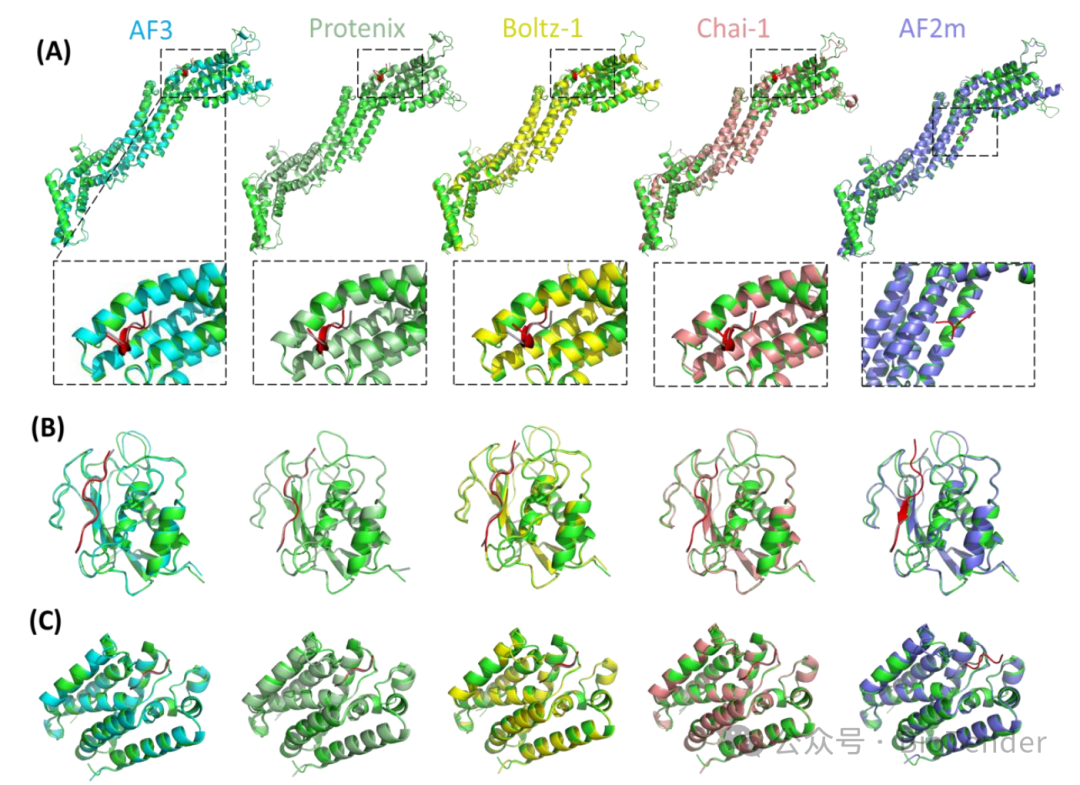
Representative cases demonstrating the superiority of next-generation modeling methods over previous-generation methods
Is AF3 really stronger than AF2m??
The research results show that the new generation method has demonstrated significant advantages in protein-short peptide prediction!
Comparison of Individual Methods
AF2m (AlphaFold2-multimer): The success rate of high-precision prediction is only 53%, performing the worst.
AF3 (AlphaFold3): Increased to 76.8%, showing significant improvement!
Chai-1 (with MSA): Success rate 78.8%, even without MSA, it is 70.7%.
Boltz-1: Reached 71.7%, slightly lower than Chai-1.
Protenix: The best performer, reaching 80.8%, becoming the "champion" of this evaluation!
Success Rate at Different Precision Thresholds
| Protenix | 80.8% | 90.0% | 89.9% |
It can be seen that the single model of Protenix has the best prediction effect, while the success rates of models such as AF3 and Chai-1 also far exceed AF2m.
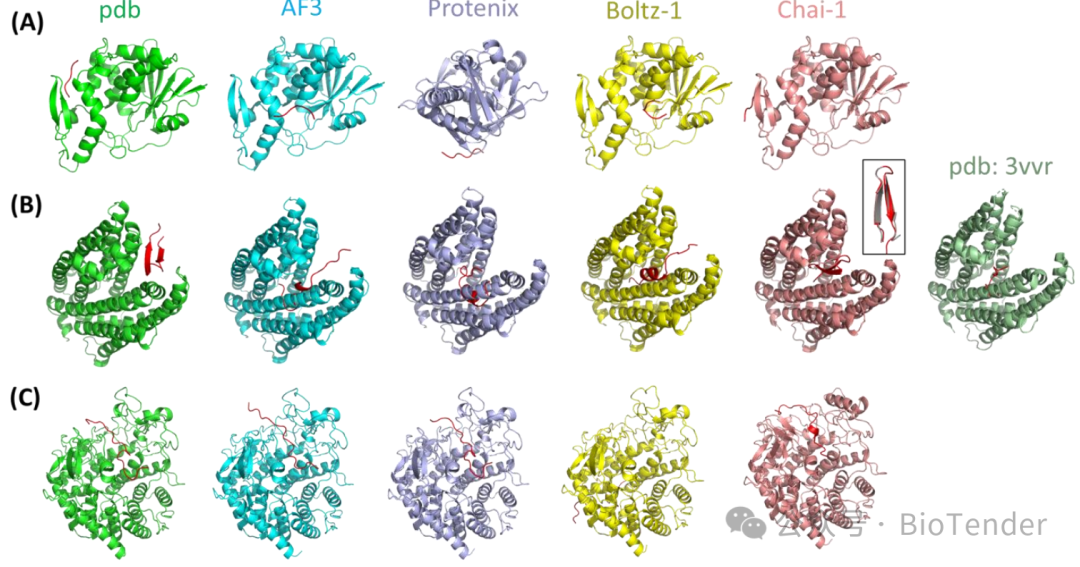
Representative cases of unsuccessful structural prediction
Combined Strategy: Two Models Work Together, Accuracy Reaches 90%!
Although individual methods have seen some improvement, the research team found thatThe effect will be better if multiple methods are combined!
Optimal Combination Strategy
AF3+Protenix Combination:
High-precision prediction success rate fromIncreased from 80% to 89%。 Medium Precision Prediction Success Rate Reaches97%。 AF3+Protenix+Chai-1 Combination
The success rate of high-precision prediction has been further improved to91%! Coverage97%The above protein-peptide complex.
Comparison of prediction accuracy of receptors and pocket sites
Why Combination Strategies Work?
The study found that different models in predictionComplementarity existsThat is, the correct prediction of certain complexes may only be successfully achieved by a specific model. For example:
Protenix independently predicted successful complexes: 7
Chai-1 successfully predicted complexes alone: 2
When AF3, Protenix, and Chai-1 are used in combination, the number of successfully predicted complexes increases to 94 (95% of 99).
This indicates that,Different models may have their own advantages in predicting different types of protein-peptide complexes, so combining them can achieve higher prediction success rates!
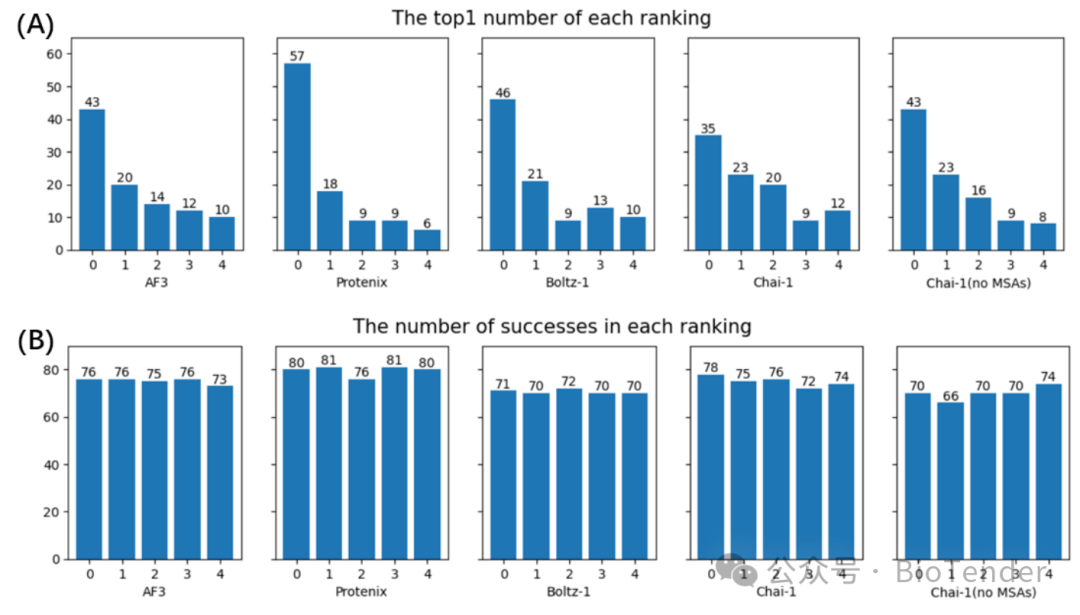
Performance of each model (from ranked 0 to ranked 4) for each method
Reasons for Prediction Failure
Despite the excellent performance of the new generation methods, there are still some failure cases, and the main reasons include:
Receptor Protein Structure Prediction Error: Some models failed to correctly model the spatial conformation of the protein.
Binding Site Prediction Error: Some models failed to identify the correct binding position of the peptide.
Binding Mode Prediction Error: The predicted short peptide binding mode deviates significantly from the actual structure.
For example, in 3N2D (Type I ribosome-inactivating protein complex)In the prediction, none of the methods were able to correctly predict the binding sites of the short peptide. Meanwhile, inIn the prediction of 3WBN (MATE transporter complex), the model failed to recognize the characteristic of the short peptide being a cyclic peptide, leading to an incorrect binding mode.
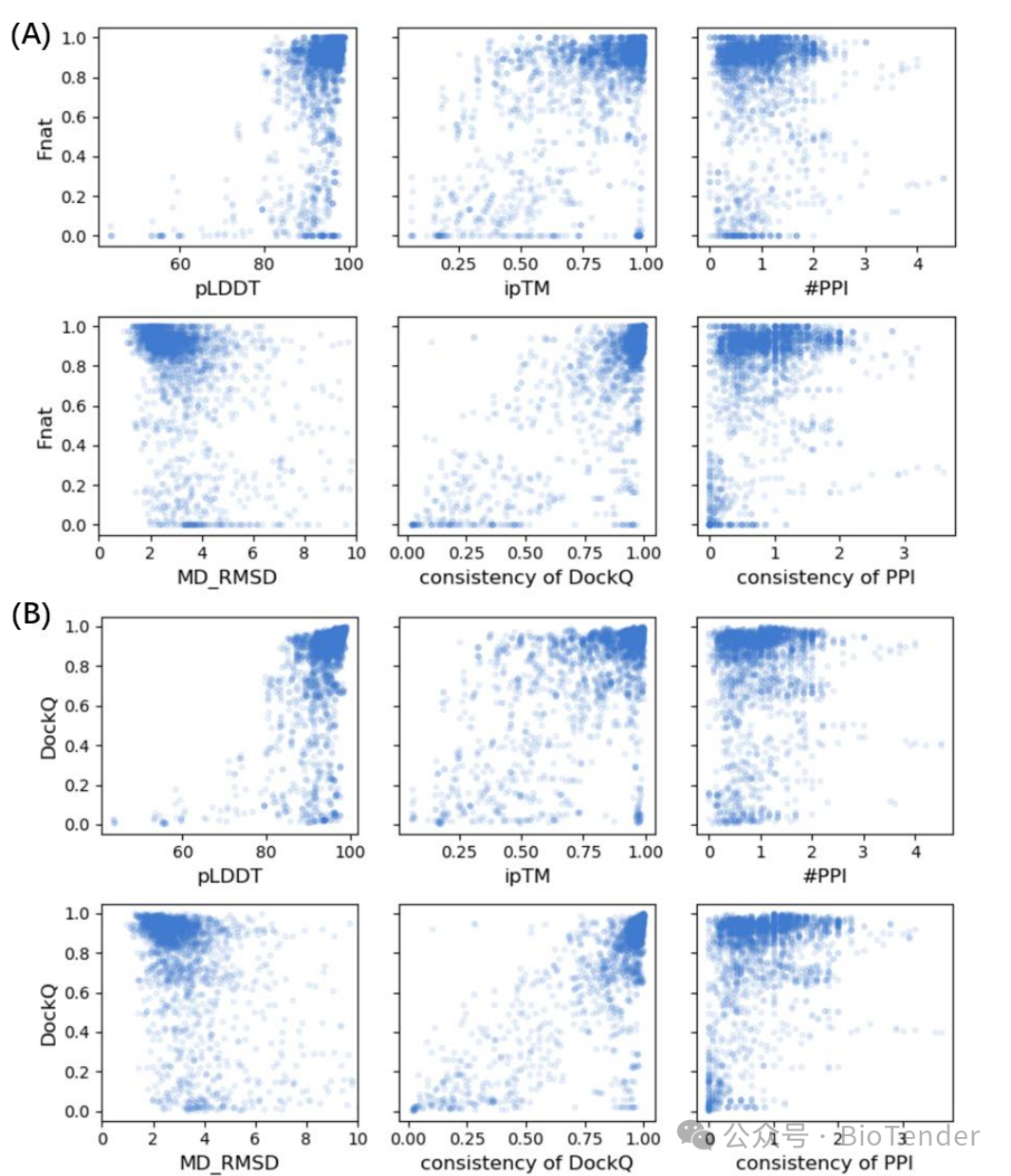
Correlation of different indicators with (A) Fnat and (B) DockQ
Significance and Prospects of the Study
This study is the firstThe system evaluated the performance of AF3 and its derivative methods in predicting protein-short peptide complexes., and proposed an effectiveMulti-Model Combination Strategy, increasing the high-precision prediction success rate toMore than 90%. This not only providesProtein Interaction ResearchProvided new tools, and also forShort Peptide Drug DesignHas brought more precise calculation methods.
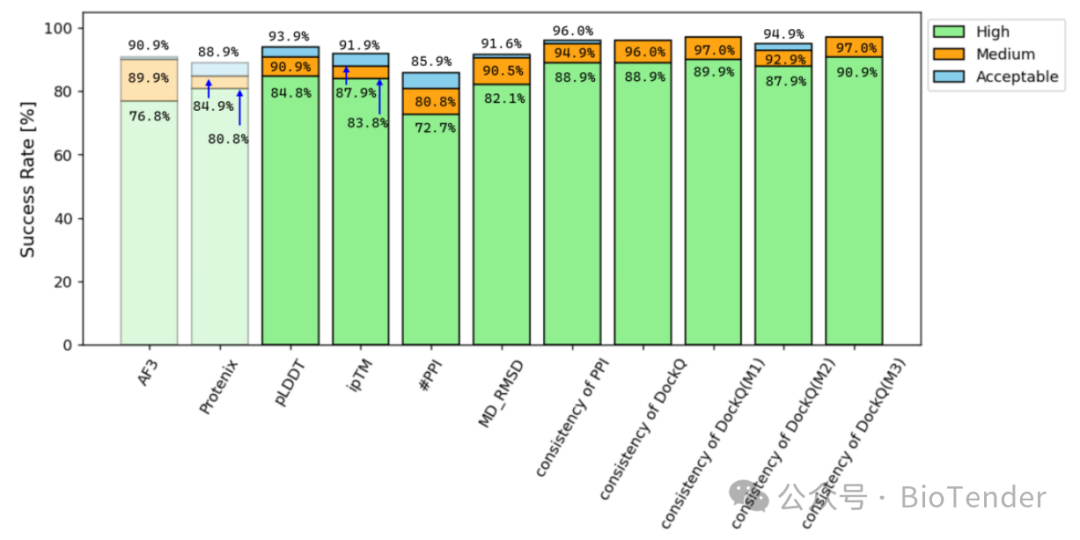
Success rates of protein-peptide structural predictions after filtering methods/models with indicators
In the future, this field may continue to develop in the following directions:
Optimize AI models based on experimental data to further improve prediction accuracy.
Develop smarter predictive screening algorithms to reduce computational resource consumption and improve efficiency.
Expanding to protein-small molecule and protein-nucleic acid interaction prediction, promoting the application of AI in structural biology.
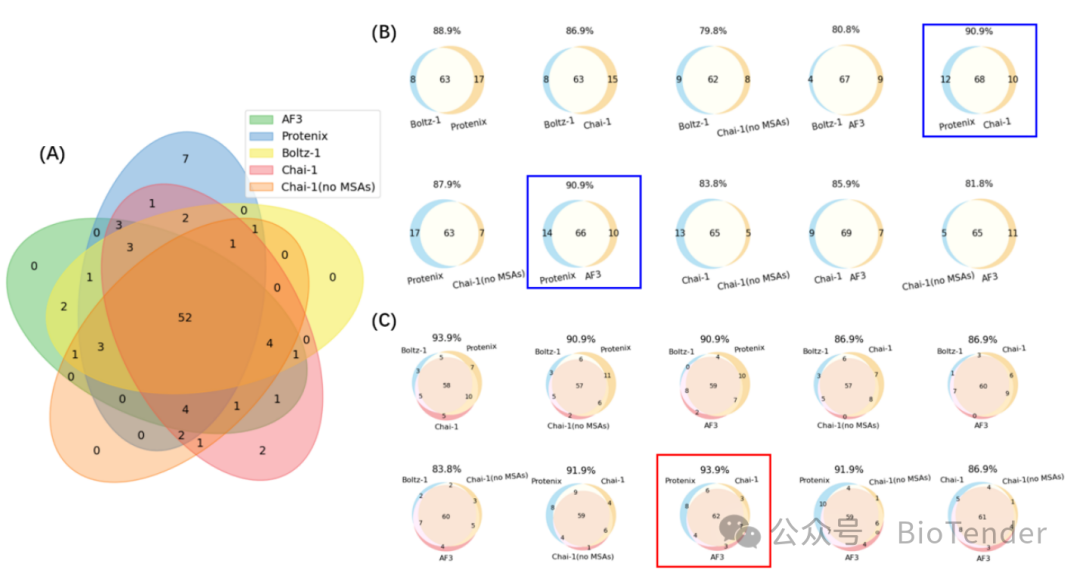
Overlap of successful cases predicted by different modeling methods
The new generation of AI models is accelerating the unraveling of the mysteries of biomacromolecules. More breakthroughs are expected to boost precision medicine and new drug development!
Reprinted from: BioTender
About Zelixir
