
High-quality molecular representations are crucial for AI-driven drug discovery. Although Graph Neural Networks (GNNs) have made some progress in this field, challenges such as limited labeled molecules, data imbalance, and overfitting still persist. Augmentation techniques have become a mainstream solution, but directly modifying the topology of molecular graphs may lead to the loss of critical information. Meanwhile, due to the sparsity and complexity of molecular data, adversarial augmentation is also prone to introducing noise.
Recently, the collaborative paper "Adaptive symmetry-based adversarial perturbation augmentation for molecular graph representations with dual-fusion attention information" by MindRank, Wuhan University of Science and Technology, Xiamen University, and Hunan University teams was published in the top international journal in the field of information fusion, Information Fusion (IF: 14.8).The paper proposes a new plug-and-play architecture called GapCL, which introduces a symmetric perturbation mechanism in gradient-based adversarial enhancement to preserve critical chemical space information. Meanwhile, GapCL integrates a dual attention mechanism to strengthen key information and combines contrastive learning to achieve adaptive perturbation strategies.In 12 molecular property prediction tasks, GapCL significantly enhanced the model's robustness and generalization ability.Model performance reachesBest in class.The experimental results show that this method has leading performance and can effectively enhance the molecular graph representation capability.

1. Introduction
In recent years, artificial intelligence has made remarkable progress in multiple fields. Deep learning, due to its powerful capabilities, has gained widespread attention in areas such as drug discovery. Among these, molecular representation models play a central role in tasks like drug design, property prediction, and virtual screening by converting molecular structures into vectors or features that computers can process, capturing key information such as chemical properties, geometric configurations, and electron distributions, thereby becoming an essential support for precise drug discovery.
Considering the complexity of molecular structures and the diversity of compounds, constructing high-quality molecular representations requires more powerful modeling methods. Compared to traditional methods that rely on manual features or shallow learning, Graph Neural Networks (GNNs) can more effectively extract topological information from graph structures composed of atoms and bonds, adapting to various downstream tasks. In this paper, researchers temporarily classify graph-structured Transformers as a type of GNN model to discuss their advantages in molecular representation from a unified perspective.
Despite the many advances in GNN, its generalization and robustness are still limited by the scarcity of labeled molecular data. Existing graph augmentation methods can alleviate the insufficiency of labeling but may disrupt the chemical properties of molecules. Moreover, most methods lack feature correction after perturbation, increasing application complexity and cost.
For this reason,Researchers Propose GapCL Architecture, Combining Gradient-Based Adversarial Perturbations with a Contrastive Learning Strategy that Fuses Dual Attention Information.Unlike directly modifying the graph structure, GapCL introduces adversarial perturbations in the node feature space and divides symmetric orbits based on graph automorphism, applying mean perturbations to nodes within the orbits to preserve structural consistency. Meanwhile, through a dual-fusion attention module, it adaptively adjusts the perturbation intensity during training, enhancing key information and suppressing irrelevant noise to achieve more refined molecular representations.
Under strict structural skeleton division, researchers tested five representative GNN models and two adversarial learning methods on 12 tasks from the MoleculeNet benchmark. The results showed,GapCL improves model performance in all tasks and outperforms existing adversarial enhancement methods.The visualization results further validate that GapCL enhances the discriminability of the representations.
In summary, the GapCL proposed by the researchers provides a robust, versatile, and scalable high-quality molecular graph representation learning solution. The main contributions include:
• Proposed the GapCL architecture, which can be integrated into any molecular graph representation model, combining adversarial perturbation with contrastive learning and implementing an adaptive enhancement strategy;
• Design a perturbation scheme based on symmetry and introduce a fusion attention mechanism to enhance model generalization;
• Multiple benchmark experiments validate that GapCL significantly enhances the performance of GNN models in molecular property prediction tasks.
2. Research Methods
The GapCL architecture proposed in this paper includes two key modules: the Symmetric Adversarial Perturbation Module and the Dual-Fusion Enhanced Contrastive Learning Module. The overall process is as follows:
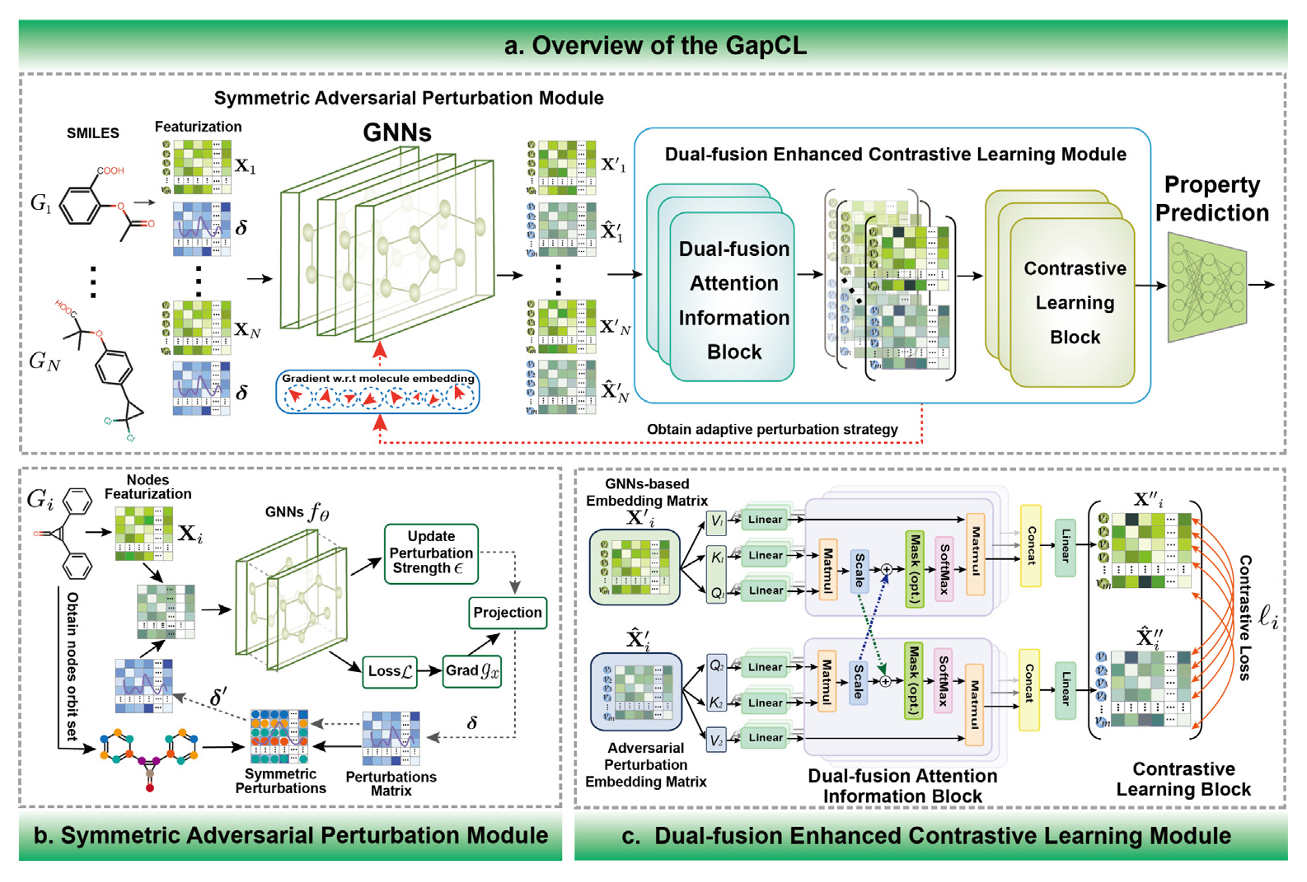
Figure 1. Schematic diagram of the GapCL architecture
Symmetric Adversarial Perturbation Module
To avoid potential loss of chemical information caused by directly modifying molecular graph topology, researchers introduced adversarial perturbations in the node feature space. Traditional augmentation methods such as deleting nodes or edges may disrupt structural consistency, whereas adversarial perturbations can enhance model robustness without altering the graph structure.
Considering the frequent presence of symmetry in molecular graphs (e.g., benzene ring structures), researchers partition nodes into orbits via graph automorphism and apply the same averaged perturbation to nodes within the same orbit. This symmetric perturbation helps maintain consistency in structural properties while enhancing diversity, thereby improving the model’s generalization ability.
Dual Fusion Enhanced Contrast Learning Module
This module aims to highlight key information and suppress irrelevant noise. First, graph neural networks extract raw node embeddings (e.g., GCN, GAT, MPNN, etc.), which are then combined with embeddings after adversarial perturbation. Feature fusion is achieved through a cross-attention mechanism, enhancing structural information and sensitive regions within molecules. The attention mechanism integrates the original and perturbed features into a unified representation using bias-weighted methods.
Subsequently, the fused features are fed into a multi-round contrastive learning module and trained using the contrastive loss (NT-Xent loss) from the SimCLR framework. By distinguishing between positive and negative sample pairs, the model can capture finer structural differences and obtain more discriminative molecular representations.
3. Experiments and Results
Dataset and Evaluation Settings
The researchers selected 12 tasks (including 9 classification tasks and 3 regression tasks) from MoleculeNet to comprehensively evaluate the performance of GapCL. All datasets were divided using a strict scaffold split, with an 8:1:1 ratio for the training set, validation set, and test set.
Classification tasks use ROC-AUC as the evaluation metric, and regression tasks use RMSE to measure error.
Baseline Model and Comparative Methods
• Graph Representation Models: Including GCN, GAT, MPNN, CoMPT, Uni-mol.
• Adversarial Learning Methods: Including PGD and FLAG Two Enhancement Methods.
• Other comparative models: including D-MPNN, Attentive FP, PretrainGNN, MolCLR, GROVER, GraphMVP, GEM, PremuNet and other supervised or pre-trained models.
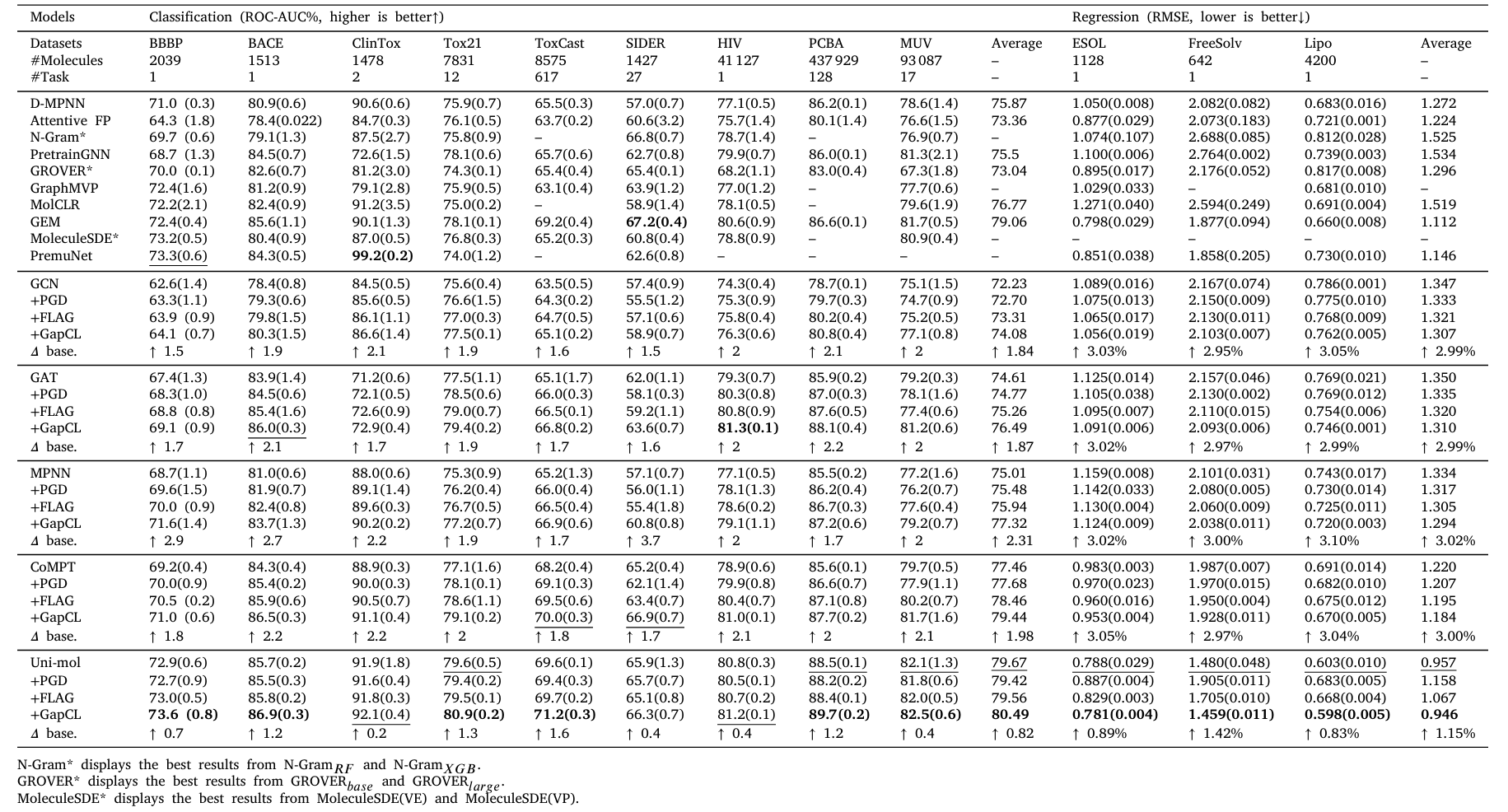
Table 1. Evaluation results of different methods on 9 classification and 3 regression datasets
Main Results
GapCL can comprehensively enhance the performance of all base models, and it also outperforms PGD and FLAG. Particularly on non-pre-trained models such as GCN, GAT, MPNN, and CoMPT, GapCL achieves an average improvement of approximately 2% in classification tasks and about 3% in regression tasks. For the powerful pre-trained model Uni-mol, GapCL still provides a certain level of improvement, demonstrating the method's versatility and scalability.
Ablation Experiment
To verify the contribution of each component, the researchers removed certain modules for testing on five models. The experimental results show:
• Using symmetric perturbation alone (+AP) has a certain effect;
• The effect is slightly improved by combining the dual-fusion attention module (+AP_DF);
• The complete GapCL (AP + DF + Contrastive Learning) significantly outperforms other combinations, indicating a notable synergistic enhancement among the modules.
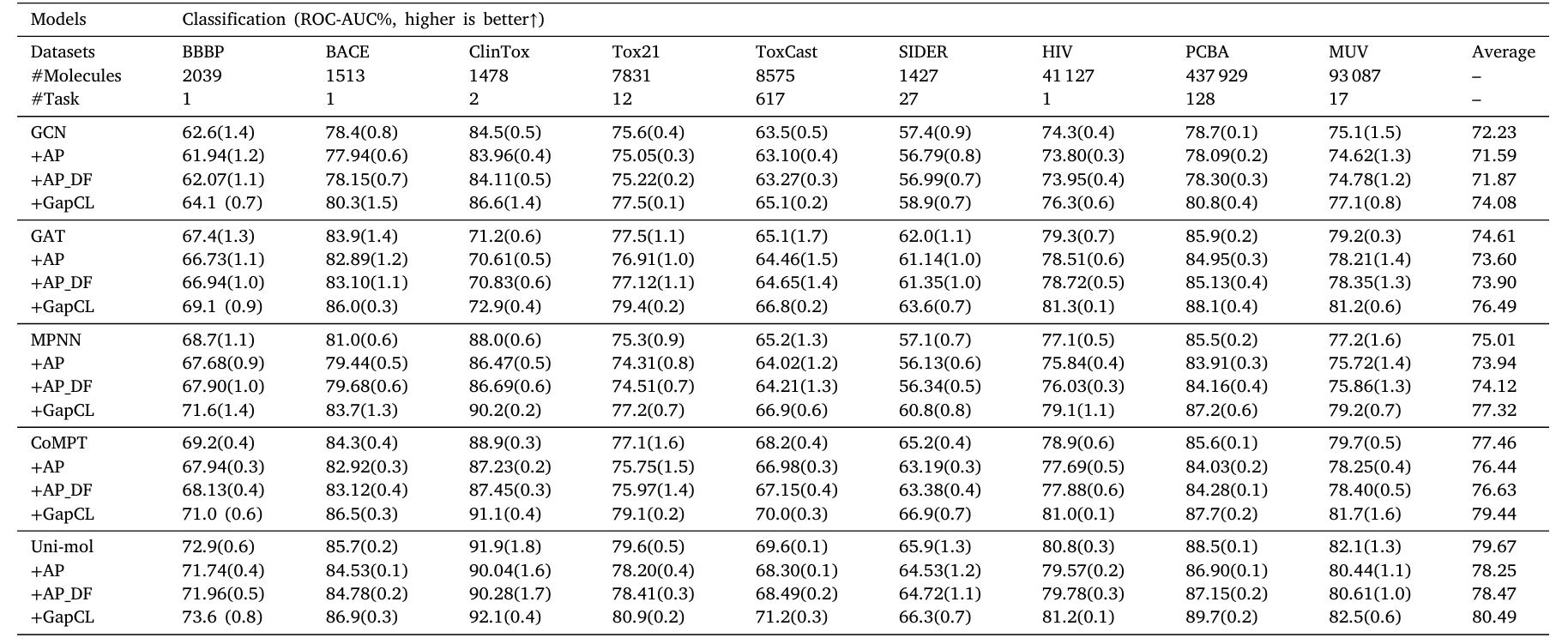
Table 2. Ablation Experiment Results
Visualization Analysis
UMAP Dimensionality Reduction Visualization on Tox21 and Lipophilicity Datasets. The model representation enhanced by GapCL is more compact, providing clearer differentiation between toxic and non-toxic molecules, as well as the trend of lipophilicity values, demonstrating stronger structural sensitivity and representation ability.
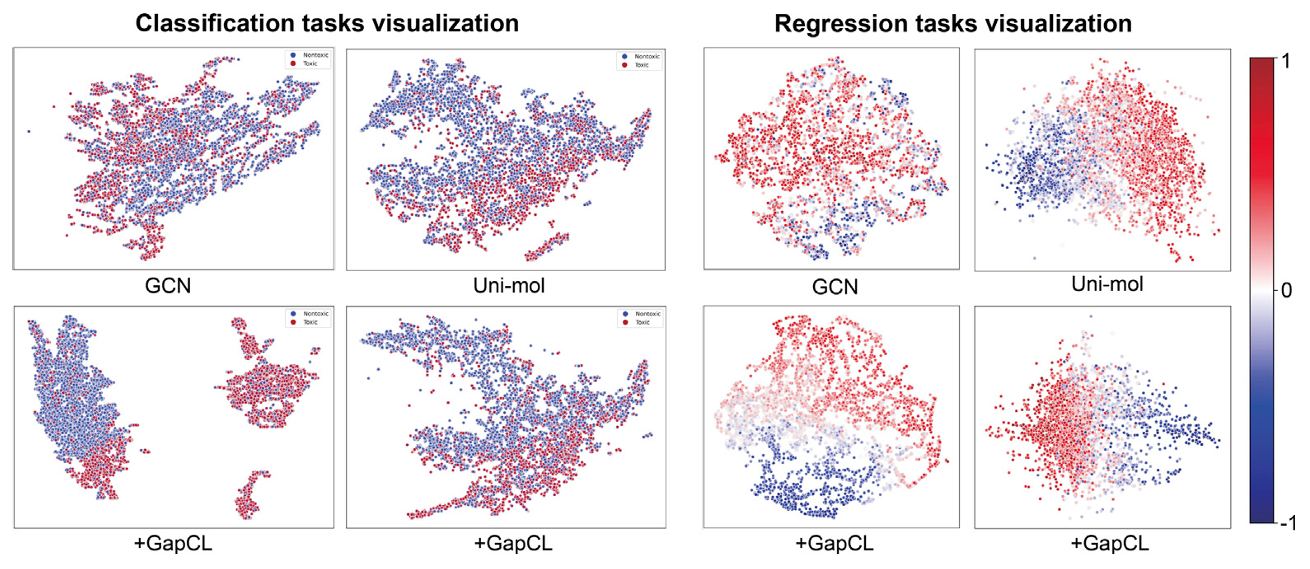
Figure 2. Visualization of the latent space through UMAP
Convergence Analysis
Taking the BBBP dataset as an example, GapCL significantly accelerates model convergence and achieves lower loss values in the early stages of training, indicating a more stable and efficient optimization process.
Hyperparameter Sensitivity Analysis
Researchers evaluated the impact of perturbation intensity ε and iteration rounds M on model performance. The results showed that the model achieved optimal performance when ε=0.001 and M=3. The best performance was also obtained when the contrastive learning loss weight was set to 0.15. Therefore, this combination was adopted as the default parameter configuration in the experiment.
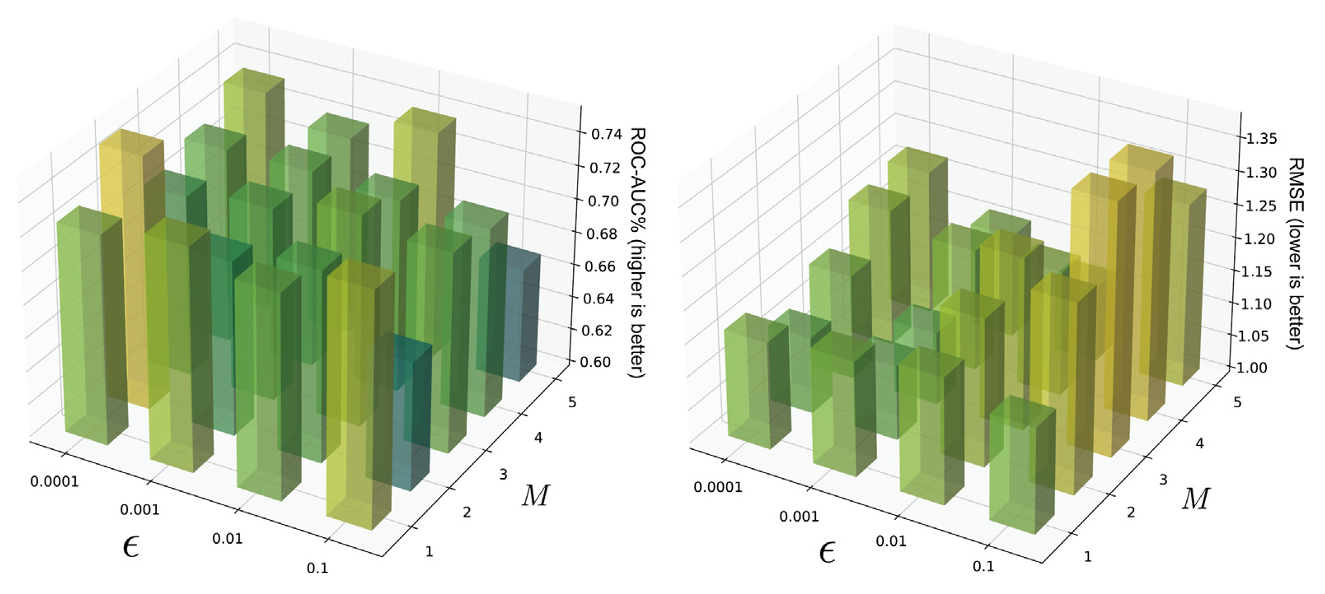
Figure 3. Hyperparameter Sensitivity Analysis on BBBP and ESOL Tasks
4. Discussion and Conclusion
This paper proposes a novel plug-and-play architecture GapCL, which combines a gradient-guided symmetry adversarial perturbation mechanism with a dual-fusion enhanced contrastive learning strategy.To enhance the robustness and generalization ability of graph neural networks in molecular graph representation learning, especially excelling in scenarios with limited annotated data.
Experiments on the MoleculeNet benchmark have demonstrated that models equipped with GapCL significantly enhance the quality of molecular representations, further advancing their application in...Drug Discovery, Material DesignThe application potential in tasks such as dependency parsing of graph-structured data.
The advantage of GapCL lies in its flexible structure and strong versatility.Can be integrated as an enhancement module into any existing GNN framework,Help the model extract key features more effectively and enhance classification and prediction capabilities. Despite the significant effect of GapCL, its method still incurs a certain computational cost, mainly due to the introduction of symmetric perturbation and dual attention fusion, which slightly increases training time. However, experiments show that this additional cost is manageable in standard GNN processes, and the performance gains far outweigh the computational burden.
Moreover, the current method still has potential for further performance improvement. Future research can explore lighter perturbation strategies, cross-task transferability, or extend its application to non-molecular graph domains to generate more robust and transferable high-quality graph representations.
In summary,GapCL provides a robust and scalable new direction for high-quality molecular graph representation learning.This also expands the methodological boundaries for the application of graph neural networks in scientific problems.
Paper Link:https://doi.org/10.1016/j.inffus.2025.103062