|Edited by the Content Team of Zhong Peptide Biochemical
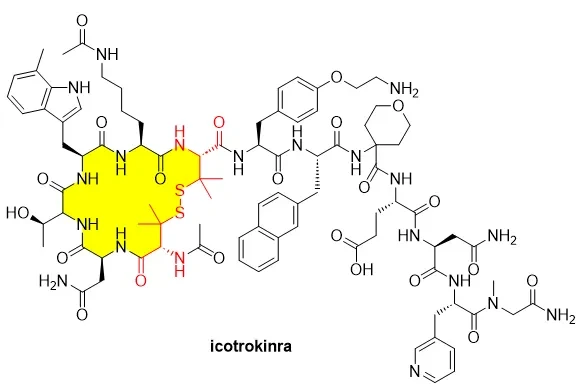
Johnson & Johnson recently announced the latest data from its Phase III clinical trial (ICONIC-LEAD) for icotrokinra (Figure 1), a peptide asset, in patients with severe plaque psoriasis. Nearly half of the patients with moderate to severe plaque psoriasis achieved complete clearance by Week 24. Additionally, another Phase III study, ICONIC-ADVANCE 1&2, showed that icotrokinra outperformed deucravacitinib in psoriasis treatment. Based on these positive results, Protagonist and Johnson & Johnson plan to initiate the world’s first head-to-head study (ICONIC-ASCEND) comparing the efficacy of oral icotrokinra with injectable biologic ustekinumab. This head-to-head study will further clarify icotrokinra's position in psoriasis treatment, particularly its advantages in terms of oral convenience and superior efficacy compared to biologics.Figure 1. Chemical structure of Icotrokinra.Icotrokinra is an oral targeted peptide drug designed to inhibit the IL-23 signaling pathway by blocking the interleukin-23 receptor (IL-23R). IL-23 is a well-known pro-inflammatory cytokine that plays a key role in immune-mediated skin diseases such as psoriasis. Traditionally, the IL-23 pathway has been a target for many biologics, and the advantage of icotrokinra lies in its nature as an oral peptide drug, offering a more convenient treatment option compared to existing subcutaneous or intravenous injections.Icotrokinra is expected to become the first approved oral IL-23 receptor inhibitor, marking a revolutionary advancement in psoriasis treatment. Many potent treatment options currently on the market, such as adalimumab (Humira) and etanercept (Enbrel), require subcutaneous or intravenous injection, potentially impacting patient compliance. Icotrokinra, administered orally, offers a more convenient treatment option compared to these injectable drugs, significantly improving patients' quality of life. Johnson & Johnson also plans to expand the use of icotrokinra to treat other immune-mediated diseases, such as ulcerative colitis and Crohn's disease, further validating its broad application potential in the field of immune modulation.Notably, Icotrokinra is a cyclic peptide containing disulfide bonds, and the development of such cyclic peptides as oral formulations has been relatively rare in the history of pharmaceutical development. Unlike conventional disulfide-bonded peptides such as oxytocin and atosiban, the disulfide bond in the structure of Icotrokinra does not originate from two cysteines but is instead formed through penicillamine (Pen). Penicillamine (Pen) is an analog of cysteine with two methyl groups attached to its C3 atom, thus it can be considered as 3,3-dimethylcysteine (Figure 2). Penicillamine is a natural degradation product of β-lactam antibiotics, penicillins.Figure 2. Chemical structure of L-penicillamine.D-Penicillamine has already been approved by the FDA and marketed as a drug under the brand name Cuprimine. It is used to treat Wilson's disease (a rare genetic disorder of copper metabolism) and is also used in patients with high urinary cystine levels, rheumatoid arthritis, kidney stones caused by various heavy metal poisonings, and scleroderma. Notably, D-(S)-penicillamine is therapeutically active, while L-(R)-penicillamine is toxic because it inhibits the action of pyridoxine (vitamin B6). The amino acid component appearing in icotrokinra is L-(R)-penicillamine.In addition to icotrokinra, an increasing number of peptide drug designs incorporate penicillamine (Pen) as a substitute amino acid for cysteine, endowing the peptide drugs with unique pharmacokinetic and pharmacodynamic properties.A research report describes the protein design of novel metallopeptides that mimic the binding loop of nickel-containing superoxide dismutase (NiSOD). In the study, D-penicillamine was introduced into the peptide chain, synthesizing the H(Pen)DLPCGLY (wtPen) peptide. Its structure exhibits a well-defined α-helical folding state. This peptide derivative demonstrates excellent superoxide dismutase activity. The penicillamine structure is located near the catalytic center, and its presence prolongs the half-life of nickel(III). In contrast, in the dismutation reaction catalyzed by natural NiSOD enzyme fragments, nickel(III) exists only at relatively low concentrations.In a study on α-Conotoxins, researchers replaced cysteine in the natural compound with Pen to develop non-opioid analgesics targeting nicotinic acetylcholine receptors (nAChRs). By selective Pen substitution and replacement with both natural and unnatural amino acids, the researchers synthesized analogs of α-Conotoxin RgIA, resulting in a peptide, RgIA-5474 (Figure 3), with 9000-fold increased activity at the human α9α10 nAChR and significantly reduced degradation caused by disulfide scrambling. This stabilized peptide potently blocks α9α10 nAChR without affecting opioid or other pain-related targets. Moreover, RgIA-5474 effectively reverses chemotherapy-induced neuropathic pain.Figure 3. Development process of RgIA-5474, an analog peptide of α-conotoxin RgIA containing penicillamine residues. (Image source: J Med Chem)In another study, researchers developed a CXCR4 antagonist (a molecule that blocks the function of the chemokine receptor CXCR4 (C-X-C Motif Chemokine Receptor 4). CXCR4 is a G protein-coupled receptor that plays a key role in immune regulation, hematopoiesis, and cancer progression). They created a highly potent, selective, and plasma-stable cyclic peptide, Ac-Arg-Ala-[d-Cys-Arg-Phe-Phe-Cys]-COOH (with a disulfide bond formed between d-Cys and Cys), but its activity (IC₅₀≈53 nM) was still insufficient for preclinical studies. To address this, the researchers developed an optimized analog, Ac-Arg-Ala-[d-Cys-Arg-Phe-His-Pen]-COOH. In this cyclic peptide, Pen replaced Cys in the original molecule and was linked to d-Cys via a disulfide bond, while the aromatic amino acid residues in the original cyclic peptide were also substituted. The optimized cyclic peptide achieved subnanomolar affinity for CXCR4 and exhibited high selectivity for CXCR3 and CXCR7 (Figure 4). Biological experiments showed that this Pen cyclic peptide inhibited CXCL12-mediated cell migration and CXCR4 internalization more effectively than plerixafor (trade name Mozobil), an approved CXCR4 antagonist. Its in vivo stability was also significantly improved compared to the earlier version of the cyclic peptide.'%20fill='%23FFFFFF'%3E%3Crect%20x='249'%20y='126'%20width='1'%20height='1'%3E%3C/rect%3E%3C/g%3E%3C/g%3E%3C/svg%3E) Figure 4. Pen-substituted Cys disulfide cyclic peptide CXCR4 antagonist. (Image source: J. Med. Chem.)From the perspective of chemical synthesis, researchers have found that the reaction rate of forming heterodisulfide bonds between Pen and Cys is faster than that of forming homodisulfide bonds between Pen-Pen or Cys-Cys. Moreover, Pen is highly prone to undergo thiol-disulfide exchange reactions with Cys-Cys, producing Pen-Cys heterodisulfide bonds. Due to the steric hindrance effect of Pen, the tendency of this heterodisulfide bond to undergo thiol-disulfide exchange is much lower than that of Cys-Cys homodisulfide bonds. This discovery provides a basis for the orthogonal reaction of Pen-Cys disulfide bond formation and the design of disulfide-containing peptide drugs. In addition, researchers designed a 23-residue linear peptide containing 2xCys and 2xPen, which, through air oxidation cyclization, yielded a high proportion of isomers of two Cys-Pen bicyclic peptides, while the third isomer of Cys-Cys and Pen-Pen did not form.Pen provides enhanced chemical stability to disulfide bonds, potentially laying the groundwork for the oral delivery of cyclic peptides with disulfide bonds, such as icotrokinra. The two methyl groups on the Cβ of the Pen side chain can hinder the attack of nucleophiles on the sulfur atom also located on Cβ, thereby enhancing the chemical stability of what would otherwise be an unstable disulfide bond. In one study, researchers replaced the disulfide bond with a thioether bond between Pen and Abu (aminobutyric acid), significantly improving the chemical stability of the original disulfide-containing peptide (Figure 5).Figure 5. Chemical structure of Pen-Abu cyclic peptide. (Image source: Org. Biomol. Chem.)Other polypeptide molecules containing Pen residues:
Figure 4. Pen-substituted Cys disulfide cyclic peptide CXCR4 antagonist. (Image source: J. Med. Chem.)From the perspective of chemical synthesis, researchers have found that the reaction rate of forming heterodisulfide bonds between Pen and Cys is faster than that of forming homodisulfide bonds between Pen-Pen or Cys-Cys. Moreover, Pen is highly prone to undergo thiol-disulfide exchange reactions with Cys-Cys, producing Pen-Cys heterodisulfide bonds. Due to the steric hindrance effect of Pen, the tendency of this heterodisulfide bond to undergo thiol-disulfide exchange is much lower than that of Cys-Cys homodisulfide bonds. This discovery provides a basis for the orthogonal reaction of Pen-Cys disulfide bond formation and the design of disulfide-containing peptide drugs. In addition, researchers designed a 23-residue linear peptide containing 2xCys and 2xPen, which, through air oxidation cyclization, yielded a high proportion of isomers of two Cys-Pen bicyclic peptides, while the third isomer of Cys-Cys and Pen-Pen did not form.Pen provides enhanced chemical stability to disulfide bonds, potentially laying the groundwork for the oral delivery of cyclic peptides with disulfide bonds, such as icotrokinra. The two methyl groups on the Cβ of the Pen side chain can hinder the attack of nucleophiles on the sulfur atom also located on Cβ, thereby enhancing the chemical stability of what would otherwise be an unstable disulfide bond. In one study, researchers replaced the disulfide bond with a thioether bond between Pen and Abu (aminobutyric acid), significantly improving the chemical stability of the original disulfide-containing peptide (Figure 5).Figure 5. Chemical structure of Pen-Abu cyclic peptide. (Image source: Org. Biomol. Chem.)Other polypeptide molecules containing Pen residues:- DPDPE([D-Pen2,D-Pen5]Enkephalin)
DPDPE is a synthetic opioid peptide and a selective agonist of the δ-opioid receptor (DOR). It was developed in the early 1980s and was the first highly selective DOR agonist to be developed. DPDPE is derived from structural modifications of methionine enkephalin.SKF 106760 is a potent platelet fibrinogen receptor (glycoprotein IIb/IIIa) antagonist with significant antiplatelet and antithrombotic activity. SKF 106760 inhibits platelet aggregation in dogs in vitro and prevents thrombus formation in stenotic carotid arteries. SKF 106760 also demonstrates the ability to eliminate aspirin-resistant thrombi formed in highly stenotic environments.Urantide is a selective and competitive urotensin-II (UT) receptor antagonistic peptide that blocks human urotensin-II (hU-II)-induced aortic contraction in rat models. Urantide can be used to study the pathological role of hU-II in the mammalian cardiovascular system.- World Intellectual Property Organization, WO2014165449 A1 2014-10-09 (CAS No. 1629641-51-5)
Inhibition of α4β7 binding to MAdCAM (mucosal addressin cell adhesion molecule) with high selectivity, without affecting α4β1 binding. This α4β7 antagonist monomer peptide can be used as an anti-inflammatory and immunosuppressant for the treatment of diseases associated with α4β7-mediated tissue function, with potential indications including gastrointestinal diseases and other inflammatory conditions.- World Intellectual Property Organization, WO2020225095 A1 2020-11-12 (CAS No. 2549162-21-0), Bayer Patent
Mannose-binding lectin (MBL)-associated serine protease (MASP) inhibitory peptides, used alone or in combination for the treatment or prevention of kidney and cardiovascular diseases as well as ischemia-reperfusion injury.- World Intellectual Property Organization, WO2008142517 A2 2008-11-27 (CAS No. 1084788-93-1), Johnson & Johnson Patent
(Ac-Pen-Lys-Pro-Val-NH2)2, used for the prevention or treatment of microbial infections. It has anti-inflammatory, fibroblast proliferation, interleukin-8 induced neutrophil chemotaxis, and candidacidal effects.- World Intellectual Property Organization, WO2024163643 A1 2024-08-08 (CAS No. 3052905-80-0)
Peptide Inhibitor of Interleukin-23 Receptor (IL-23R). Can be used for the treatment of autoimmune inflammatory diseases and related conditions.
[1] Bonczidai-Kelemen, D. et al. Introducing the penicillamine moiety into a metallopeptide mimicking the NiSOD enzyme: electronic and kinetic effects. Inorg. Chem. Front. 2022, 9, 310-322
[2] Gajewiak, J. et al. Selective Penicillamine Substitution Enables Development of a Potent Analgesic Peptide that Acts Through a Non-Opioid Based Mechanism. J. Med. Chem. 2021, 64, 9271–9278.
[3] Di Maro, S. et al. Structure–Activity Relationships and Biological Characterization of a Novel, Potent, and Serum Stable C-X-C Chemokine Receptor Type 4 (CXCR4) Antagonist. J. Med. Chem. 2017, 60, 9641–9652.
[4] Zheng, Y. et al. Orthogonal Cysteine–Penicillamine Disulfide Pairing for Directing the Oxidative Folding of Peptides. J. Am. Chem. Soc. 2015, 137, 48, 15094–15097.
[5] Duan, Z. et al. Triscysteine disulfide-directing motifs enabling design and discovery of multicyclic peptide binders. Nat Commun 15, 7799 (2024).
[6] Cui, J-B. et al. Chemical synthesis of disulfide surrogate peptides by using beta-carbon dimethyl modified diaminodiacids. Org. Biomol. Chem. 2021, 19, 9021