Insight Weekly Drug Report: 9 New Drugs/New Indications Approved in China; AstraZeneca, GSK, Sanofi Release H1 Earnings

GSK
Pharmaceutical R&D Manufacturer

Sanofi
Pharmaceutical Manufacturer

Ascendis Pharma
Disease Treatment Drug Developer

According to statistics from the Insight database, this week (July 27 - August 2), a total of 91 innovative drugs (including improved new drugs) worldwide advanced to new stages of development, with 3 receiving approval for marketing and 2...24 products have initiated clinical trials, and 20 products have applied for clinical trials.
Below, Insight will introduce the progress of some key projects at home and abroad this week.


On July 28, 2025, local time, Ascendis Pharma announced that the U.S. FDA had approved its long-acting growth hormone drug, TransCon hGH.(lonapegsomatropin-tcgd)Supplemental Biologics License Application(sBLA),For the treatment of adult growth hormone deficiency(GHD)。
Previously, the drug had received FDA approval for treatment.Children with GHD。

Screenshot source:Ascendis Official Website
TransCon hGH is a prodrug of human growth hormone developed based on the TransCon proprietary technology platform, with a unique advantage in its mechanism of action. Unlike traditional long-acting growth hormone analogs, this drug...Through a once-weekly injection, unmodified natural human growth hormone is slowly released in a controlled manner within the body.The released growth hormone is identical in structure to the endogenous hormone, and its biological activity, mechanism of action, and physiological distribution are the same as those of the daily preparations widely used in clinical practice, fundamentally ensuring safety and efficacy.
Weekly dosing regimen based on TransCon technology, the number of annual injections for patients using TransCon hGHDecreased from 365 times to 52 times, with an 86% reduction in injection days.Significantly improve treatment adherence.
The approval of the adult GHD indication was primarily based on the results of the foresiGHt trial, which aimed to compare the efficacy and safety of once-weekly TransCon hGH therapy with once-weekly placebo and once-daily positive control medication in adult GHD patients.
This study enrolled 259 GHD adult subjects aged between 23 and 80 years, who were randomly assigned to different groups in a 1:1:1 ratio. Doses were adjusted based on age and estrogen intake, and the subjects received treatment with fixed doses of TransCon hGH, placebo, or active comparator drugs. The weekly dosing of TransCon hGH was equivalent to that of the daily formulation.
Data showed that at Week 38, the TransCon hGH group reached the primary endpoint and key secondary endpoints of the study. Compared with placebo, the TransCon hGH group had a significant reduction in trunk fat from baseline and a significant increase in whole-body lean mass at Week 38. In terms of safety, it was generally well-tolerated with no treatment-related discontinuations.
Previously, TransCon hGH has been approved by the US FDA and EU EMA under the brand name Skytrofa. It is the first long-acting growth hormone in Europe and the US administered once a week, used for treating children and adolescents with GHD.
Previously, Viseon Pharmaceuticals(VISEN Pharmaceuticals)Granted exclusive authorization by Ascendis, holding the exclusive rights for the development, production, and commercialization of TransCon hGH in Greater China. In March 2024, the Chinese NMPA accepted its marketing application for the pediatric GHD indication.
On July 31 local time, LENZ Therapeutics announced itsOnce daily,New Type Presbyopia Eye DropsLNZ100 (1.44% Aceclidine)Has been approved for marketing by the U.S. FDA. Phase III CLARITY study shows that most subjects after taking LNZ100Significant improvement can be achieved within 30 minutes, and the effect can last for 10 hours.。
箕星药业 has obtained LNZ100 Exclusive rights for development and commercialization in Greater China, the drug in China IIIClinical research has been achievedPositive Topline Results。

Screenshot source: Official WeChat account of the company
Aceclidine is a small molecule acetylcholine receptor agonist., which can cause temporary pupil constriction, thereby producing significant optical effects of extending viewing distance, improving near vision and visual quality.
Previously,LENZ Therapeutics Announced Two Investigational Formulations LNZ100(Acetylcorydine)With LNZ101(Aceclidine + Brimonidine)Positive results were achieved in the Phase III multicenter, double-blind, randomized, controlled study CLARITY for the treatment of presbyopia. The study includes two six-week efficacy trials, CLARITY 1 and CLARITY 2, as well as a six-month safety trial, CLARITY 3.
The results showed that inⅢTrial on Safety and Effectiveness in the Period(CLARITY 1 and CLARITY 2)In China, LNZ100 achieved the primary endpoint and key secondary endpoints. The Day 1 results of the CLARITY 2 trial showed:71% of the subjectsImprovement of near vision by 3 lines or more within 30 minutes;71% The subjectsImprovement of 3 lines or more within 3 hours; 40% achieved continuous improvement of 3 lines or more within 10 hours.
During the four-week study period, the improvement in near vision in the CLARITY 1 and CLARITY 2 trials was reproducible and consistent. Meanwhile, LNZ100 was well-tolerated, with no serious treatment-related adverse events observed across more than 30,000 treatment days in all three CLARITY trials.
LNZ101 showed similar results in the trials, including achieving primary and secondary endpoints in CLARITY 1 and CLARITY 2, but did not demonstrate superiority over LNZ100. Based on these results,LENZ Selects LNZ100 as Its Lead Candidate。
It is worth mentioning that Jixing Pharmaceuticals has obtained the exclusive rights for the development and commercialization of LNZ100 in Greater China in April 2022, and the Phase III study JX07001 for the treatment of presbyopia in Chinese patients has also achieved positive topline results.
On July 31, Haisco announced that the U.S. FDA has acceptedHSK3486 (Cyclopropofol Injection)New Drug Marketing Application(NDA)。

Source of screenshot: Corporate announcement
HSK3486(Cyclopropofol Injection, Sishuning®)Developed by Haisco with independent intellectual property rights.Class 1 Intravenous Anesthetic Drugs, was approved for marketing in China in December 2020. It has obtained drug registration certificates for the indications of "sedation and anesthesia in non-tracheal intubation surgeries/procedures," "induction and maintenance of general anesthesia," and "sedation during intensive care" in China.
HSK3486 was approved by the FDA for an IND application in January 2021, bypassing Phase II in the United States and directly entering the pivotal Phase III clinical trial. Clinical research results from within and outside of China indicate that,HSK3486 has a rapid onset and recovery, a high success rate of anesthesia, more stable blood pressure control, and significantly reduced incidence of adverse reactions such as hypotension, respiratory depression, and injection pain., with higher satisfaction among patients and doctors.
Haisco completed all clinical studies in 2024 and finished Pre-NDA communication with the FDA, reaching an agreement on the submission content. Haisco obtained the FDA's approval for NDA submission and recently received acceptance of its marketing authorization application.

This application is based on the Phase III clinical trial MATTERHORN.Positive results of the study.The trial results were presented at the 2025 American Society of Clinical Oncology (ASCO).(ASCO)Announced at the annual meeting plenary session and simultaneously published in The New England Journal of Medicine.
In the trial, patients received neoadjuvant durvalumab before surgery.(Imfinzi)Combined chemotherapy treatment, followed by adjuvant durvalumab combined with chemotherapy after surgery, and then subsequent treatment with durvalumab monotherapy.
In a pre-planned interim analysis, compared with chemotherapy alone, patients receiving a perioperative treatment regimen based on durvalumabThe risk of disease progression, recurrence, or death was reduced by 29% in patients.(Based on EFS Hazard Ratio [HR] of 0.71; 95% CI, 0.58 - 0.86; p < 0.001)The median EFS for the durvalumab treatment group has not yet been reached, while it was 32.8 months for the control group. Among patients receiving a perioperative regimen based on durvalumab, an estimated 78.2% were event-free at one year, compared to 74.0% in the control group; the estimated event-free survival rates at 24 months were 67.4% and 58.5%, respectively, indicating that the benefits of the durvalumab-based regimen increase over time.
For the key secondary endpoint of overall survival (OS), a clear trend favoring the durvalumab-based perioperative treatment regimen was observed (HR = 0.78; 95% CI 0.62 - 0.97; p = 0.025). The trial will continue to follow up on overall survival and will conduct a formal assessment at the time of the final analysis.
In terms of safety, the safety profile of the combination therapy was consistent with the known safety characteristics of each drug, and the proportion of patients completing surgery was similar to that of the chemotherapy-alone group. The incidence of grade 3 or higher adverse events due to any cause was similar in both groups.
Based on MATTERHORNResearch, regulatory applications for the drug are currently under review in the EU, Japan, and several other countries.
On July 29 local time, AbbVie announced that an sNDA application submitted to the FDA has been accepted, for Venetoclax tablets.(VENCLEXTA®,venetoclax)Co-administration of AstraZeneca's BTK inhibitor, acalabrutinibFixed Course RegimenFor Untreated Chronic Lymphocytic Leukemia(CLL)Patient.
The application is mainly based onPivotal Phase IIIAMPLIFY Study Results. This is a randomized, global, multicenter, open-label Phase III clinical trial designed to evaluate the efficacy and safety of acalabrutinib + venetoclax ± obinutuzumab versus investigator’s choice of chemoimmunotherapy in previously untreated adult patients with CLL without del(17p) or TP53 mutations. The primary endpoint of the study isAs assessed by the Independent Review Committee (IRC)PFS of the Acalabrutinib + Venetoclax Group.
The results showed,Acalabrutinib + Venetoclax Group77% of patients were progression-free within three years.Acalabrutinib + Venetoclax + ObinutuzumabThis proportion was 83%, compared to 67% in patients treated with standard chemoimmunotherapy. The median progression-free survival (PFS) was not reached, while for chemoimmunotherapy it was 47.6 months.
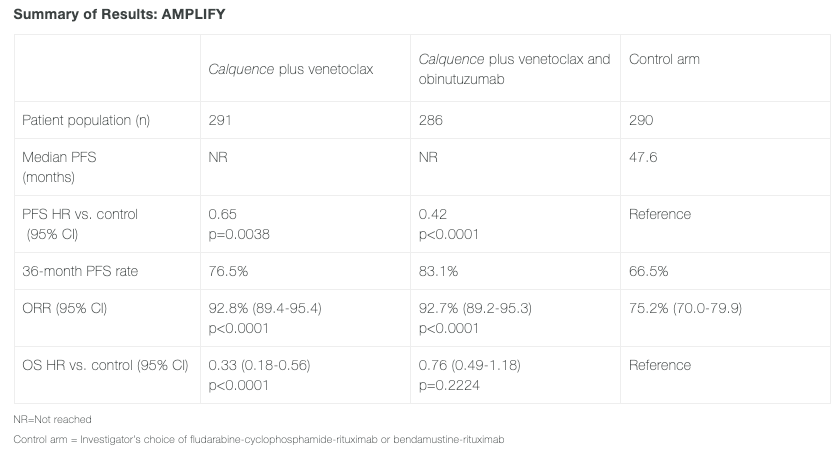
Compared with standard-of-care chemotherapy immunotherapy, acalabrutinib + venetoclax reduced the risk of disease progression or death by 35% (HR=0.65; 95%CI: 0.49-0.87; P=0.0038).Acalabrutinib + Venetoclax + Obinutuzumab reduced the risk of disease progression or death by 58% (HR 0.42; 95% CI 0.30-0.59; p<0.0001).
On July 29, AstraZeneca disclosed in its half-year financial report that it would terminateAZD5851(GPC3 CAR-T)、AZD6422(CLDN18.2 CAR-T)AndNT-125(TCR-T)The Phase I clinical development, as well as the Phase II clinical development of zibotentan + dapagliflozin for cirrhosis, and Ultomiris (ravulizumab) for lupus nephritis.

Source: AstraZeneca Official Website
It is worth noting that,After the Phase I clinical trials of the three cell therapies, AZD5851, AZD6422, and NT-125, were terminated, no related clinical trials are currently underway.
Although AstraZeneca has terminated the development of these three cell therapies, it does not mean that it is going to exit the field.Reduce layout. In the past two years, AstraZeneca has successively acquired cell therapy newcomer Gracell Biotechnologies andEsoBiotec, also withCellectis Collaborates to Develop Novel Cell Therapies.

WhenOn July 29 local time, Eli Lilly announced positive topline results from the Phase III BRUIN CLL-314 study. The trial compared non-covalent(Reversible)BTK InhibitorPitozumab(Jaypirca)Compared with the covalent BTK inhibitor Imbruvica(Ibrutinib)In chronic lymphocytic leukemia or smallLymphocytic Lymphoma(CLL/SLL) Efficacy in patients.
According to the press release,This is the first to include treatment-naive patients,Head-to-Head Phase III Study of Covalent BTK Inhibitors。

Source: Eli Lilly Official Website
The results showed that the study met its primary endpoint,PitobrutinibSuperior in terms of ORR(p <0.05)PFS has not yet matured in this analysis but has already shownPitozumabA trend of better efficacy. No impact on overall survival was observed. (OS) Any damage.
BRUIN CLL-314 Study to Be Announced SoonResults from the Phase III BRUIN CLL-313 study, which together form the basis for global regulatory submissions.BRUIN CLL-313 Study Aims to EvaluatePitozumabEfficacy of comparative chemoimmunotherapy in previously untreated CLL/SLL patients, with results expected to be announced in the second half of 2025.

At week 24, patients receiving upadacitinib 15mg and 30mg treatments36.0% and 47.1% achieved 90% or greater scalp hair coverage, respectively.(SALT≤10), whereas only 1.4% of patients receiving placebo treatment reached this endpoint.(p<0.001). Other key secondary endpoints achieved include: the percentage of subjects with improved eyebrows and eyelashes at week 24 after treatment with two doses of upadacitinib, and achieving complete scalp hair coverage (SALT=0)Percentage of subjects.
During the 24-week placebo-controlled period(Phase A)In China, the safety profiles of the two doses of upadacitinib were generally consistent with the safety profiles observed in its approved indications.
Heavyweight Pharmaceutical Deals
According to the Insight database, a total of 27 transaction events occurred this week (July 27 - August 2).
On July 28, Hengrui Medicine announced its collaboration with GSK.(GSK)Reach a cooperation agreement,The two parties will jointly develop up to 12 innovative drugs. These projects have been strictly screened,Aiming to expand GSK's established R&D pipeline in therapeutic areas such as respiratory, autoimmune and inflammation, and oncology.. This collaboration will inject strong momentum into Hengrui Medicine's globalization process, while creating significant growth opportunities for GSK post-2031.
According to the terms of the agreement, GSK will pay Hengrui Medicine a total of$500 million upfront payment。If all items receive the exercise of options and all milestones are achieved, Hengrui is expected to obtainThe total potential amount is up to approximately 12 billion US dollars.
According to the agreement, Hengrui Medicine will independently developPDE3/4 Inhibitor Innovative Drug HRS-9821The global exclusive rights, excluding mainland China, Hong Kong, Macao, and Taiwan, have been licensed to GSK for compensation.
HRS-9821 is currently in the clinical development stage and can be used for treatment.Chronic Obstructive Pulmonary Disease (COPD), as an adjunctive maintenance therapy, regardless of prior treatment regimens. The addition of HRS-9821 will strongly support GSK in achieving its strategic vision of addressing those who continue to face breathing difficulties.(Shortness of breath)Or patients who, based on their disease characteristics, are unlikely to receive inhaled corticosteroids or biologics, providing a treatment option for the widest range of COPD patient populations. In addition, HRS-9821 is also expected to be developed into a convenient dry powder inhaler.(DPI)Formulations, strategically aligned with GSK's existing inhaled product portfolio.
This agreement also includes a groundbreaking large-scale cooperation plan,Apart from HRS-9821, the two parties will jointly develop up to 11 projects.Hengrui Medicine will lead the development of these projects, up to and including Phase I clinical trials involving overseas subjects, with each project having its own financial structure. GSK will have the exclusive option to further develop and commercialize each project globally, excluding mainland China, Hong Kong, Macao, and Taiwan, by the end of Phase I clinical trials at the latest, as well as certain project substitution rights.
According to the terms of the agreement,Hengrui Medicine to Receive $500 Million in Upfront Payment, Including PDE3/4 LicenseIf all options are exercised and all milestones are achieved, Hengrui will be eligible for future milestone payments based on successful development, registration, and sales.Total potential amount approximately 12 billion US dollarsHengrui will have the right to charge GSK corresponding tiered sales royalties.(Excluding mainland China, Hong Kong, Macao and Taiwan)。
2. Upfront Payment of $130 Million! Sanofi Acquires China Rights to a Small Nucleic Acid Drug from Viya Zhen
On August 1, Sanofi announced its collaboration with Arrowhead's subsidiary, Viya Zhen.(Visirna Therapeutics)Sign an Asset Purchase AgreementViya Zhen is currently developing and commercializing four of Arrowhead's cardiovascular metabolic candidate drugs in the Greater China region. According to the agreement,Sanofi to Obtain Rights to Develop and Commercialize Plozasiran Sodium Injection in Greater China。
After the closing of the asset purchase agreement, Viya Zhen will acquire Sanofi$130 million upfront payment, and will be eligible to receive up to [amount] after the various indications for Pralsetinib Sodium are approved in mainland China.Additional milestone payments of $265 millionArrowhead is entitled to receive royalties on net sales of Lumasiran in the Greater China region.
Pulersiran Sodium, formerly known as ARO-APOC3, is a first-in-class investigational RNA interference(RNAi)Therapy Aimed at Reducing Apolipoprotein C-III(APOC3)thereby reducing triglyceride levels and restoring lipids to a more normal level. In Greater China,Plecanatide SodiumIs Familial Chylomicronemia Syndrome (FCS) and SevereHypertriglyceridemiaPotential treatment options for (SHTG).
Plegridy Sodium Injection for the treatment of FCS patients has been granted by the U.S. FDABreakthrough Therapy Designation, Orphan Drug Designation, and Fast Track Designation, as well as the Orphan Drug Designation granted by the European Medicines Agency. The marketing authorization application for the investigational drug volanesorsen sodium injection for the treatment of FCS has been submitted to multiple global regulatory agencies, but it has not yet received any approvals or authorizations for the treatment of any diseases.
3、CSPC Group and MADRIGAL Enter into Exclusive Licensing Agreement for Small Molecule GLP-1RA
On July 30, CSPC Pharmaceutical Group announced that it had entered into an exclusive licensing agreement with Madrigal Pharmaceuticals, Inc. (Madrigal) for the global development, manufacturing, and commercialization of SYH2086, an orally administered small-molecule glucagon-like peptide-1 (GLP-1) receptor agonist.
According to the terms of the agreement, CSPC has agreed to grant Madrigal an exclusive license to develop, manufacture, and commercialize SYH2086 globally, while retaining the group's rights to develop and sell other oral small-molecule GLP-1 receptor agonist products in China. The group is entitled to receive up to a total consideration of $2.075 billion, including a $120 million upfront payment, potential development, regulatory, and commercial milestone payments of up to $1.955 billion, and tiered double-digit royalties based on SYH2086's annual net sales.
SYH2086 is a preclinical candidate drug independently developed by CSPC, belonging to a new class of oral small-molecule GLP-1 receptor agonists. Preclinical data shows that SYH2086 exhibits excellent in vitro agonistic activity and significant in vivo glucose-lowering and weight-reducing effects. It also demonstrates linear pharmacokinetic (PK) behavior across a wide dose range in different animal species, with no apparent safety risks.
For detailed reporting articles, see >>AstraZeneca 2025 H1: China Revenue $3.515 Billion, Enhertu Global Sales $2.289 Billion
On July 31, Sanofi released its Q2 and H1 2025 financial report: Second Quarter, with sales of 9.94 billion euros (approximately 11 billion US dollars), an increase of 10.1%(Calculated at fixed exchange rates, same below);In the first half of the year, sales reached 19.889 billion euros (approximately 21.9 billion US dollars), increasing by 9.9%.。The exchange rate is calculated based on the average exchange rate for H1 2025, with 1 euro = 1.1004 US dollars.
By region, in the first half of the year: The U.S. market generated the highest revenue of 9.535 billion euros, increasing by 16.4%; sales in the European market reached 4.144 billion euros, growing by 1.8%; revenue in the Chinese market was 1.388 billion euros, rising by 0.1%.

For detailed coverage, see >>Sanofi Financial Report: H1 Revenue of $21.9 Billion, Dupilumab Sales Reach $8 Billion
On July 30, GlaxoSmithKline (GSK) released its H1 2025 financial report, covering the first half of the year.Revenue of 15.502 billion pounds (20.157 billion US dollars)`, up 5% year-over-year`(Calculated at fixed exchange rates, the same below)。R&D investment of 3.486 billion pounds($4.532 billion), an increase of 22% year-on-year.
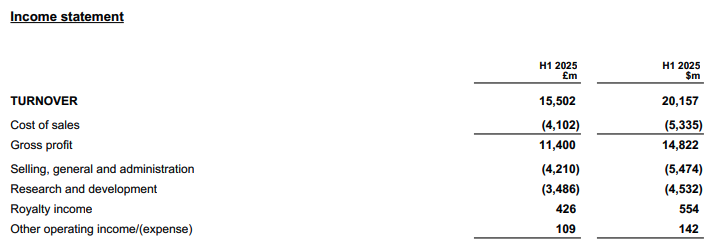
GSK's business is mainly divided intoSpecialty Medicines, Vaccines, and General MedicinesThree segments respectively contributed 62.60 billion pounds in the first half of the year.($8.141 billion, +16%)£4.186 billion($5.443 billion, +1%)£5.056 billion($6.573 billion, -3%)Performance income.
For detailed coverage, see >>GSK's Revenue Exceeds $20 Billion in the First Half of the Year, Terminates Two Phase III Studies

July 30, NMPA official website shows, Hengrui Dason「RanibizumabInjection」(HR001)Approved for marketing in China,For the treatment of adult relapsed or refractory large B-cell lymphoma after two or more lines of systemic therapy, including diffuse large B-cell lymphoma not otherwise specified, diffuse large B-cell lymphoma transformed from follicular lymphoma, high-grade B-cell lymphoma with MYC and BCL2 rearrangements, and high-grade B-cell lymphoma not otherwise specified.
Notably, this is the company's first commercially launched CAR-T cell therapy.
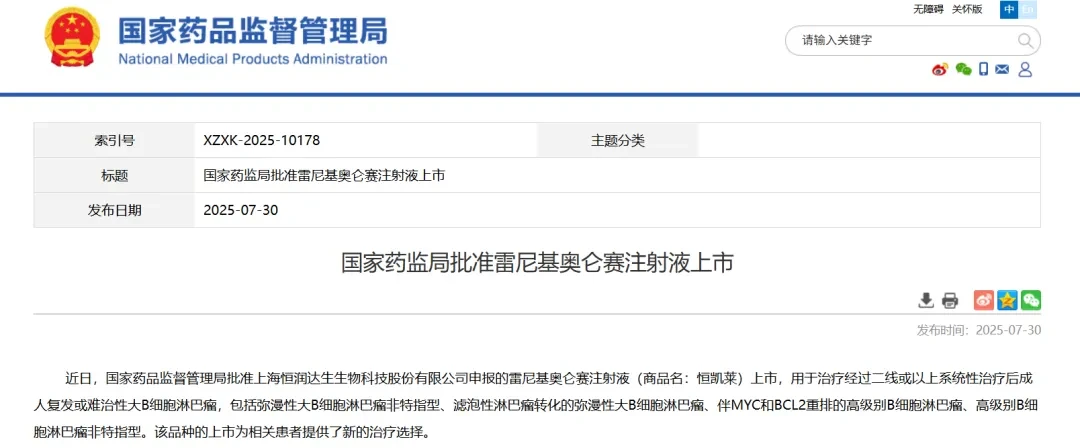
As of May 16, 2023, a total of 81 patients had received infusions. The baseline ECOG score was 0-1, 51.9% were male, and the median age was 53.4 years.(Age range: 23-74 years), 3.7% were CD19 negative, and 91.4% were diffuse large B-cell lymphoma(DLBCL)。
In terms of efficacy,
Median follow-up of 160 days(Range 7~454 days),3 Months, 6 Months, and Best Objective Response Rate(ORR)53.1%, respectively(95% CI: 41.7~64.3)、45.7%(95% CI: 34.6~57.1)And 74.1%(95% CI: 63.1~83.2);
3 Months, 6 Months, and Optimal Complete Remission Rate(CRR)32.1%, respectively(95% CI: 22.2~43.4)、29.6%(95% CI: 20.0~40.8)And 49.4%(95% CI: 38.1~60.7);
Median Time to Relief(DOR)And Progression-Free Survival(PFS)339 days respectively(95% CI: 149~NE)And 176 Days(95% CI: 91~NE), Overall Survival (OS) Not yet reached.
In terms of safety, 95.1% of patients experienced cytokine release syndrome.(CRS)Among them, 3.7% of patients experienced ≥ Grade 3 CRS, and 8.6% of patients developed Immune Effector Cell-Associated Neurotoxicity Syndrome.(ICANS),No ≥3 grade ICANS. The treatment-related mortality rate was 1.2%.
On July 28, Eli Lilly announced that the tirzepatide injection had received additional approval from China's NMPA for a new indication: to be used in conjunction with diet control and exercise.Combined Insulin(With or without oral hypoglycemic agents)Treatment, Improving Blood Glucose Control in Adult Patients with Type 2 Diabetes。
Screenshot source: Eli LillyOfficial WeChat
This new indication is primarily based on the director of the Endocrinology Department at Beijing Hospital · National Center for Geriatric Medicine.Professor Guo LixinThe SURPASS-CN-INS Study as the Principal Investigator.
The 40-week study enrolled 257 Chinese participants with T2DM.(Baseline mean age 56.7 years; duration of diabetes 12.1 years; HbA1c 8.71%; BMI 27.1 kg/m2), aiming to evaluate the efficacy and safety of tirzepatide 5 mg, 10 mg, or 15 mg compared with placebo in Chinese T2DM participants receiving basal insulin alone or in combination with metformin, with or without SGLT-2i.
SURPASS-CN-INS StudyAchieved the primary endpoint and all key secondary endpoints: At week 40, the three dose groups of tirzepatide showed significantly greater reductions in HbA1c, body weight, and fasting blood glucose from baseline compared to the placebo group; additionally, the proportion of participants achieving HbA1c targets in the tirzepatide 10 mg and 15 mg groups was also significantly higher than in the placebo group.
The safety profile of tirzepatide was consistent with previous SURPASS series studies, with the most common adverse events being gastrointestinal adverse events, mostly mild to moderate in severity. The incidence of hypoglycemia in the tirzepatide group was similar to that in the placebo group, and no severe hypoglycemic events were reported in the study.
As of now, tirzepatide has been approved for multiple indications in China, includingBlood Glucose Control, Long-Term Weight Management, and Obstructive Sleep Apnea in Adults with Type 2 Diabetes。
On July 28, Bristol-Myers Squibb announced Opdivo(Nivolumab Injection)United Yiyue(Ipilimumab Injection)The solution has been approved by China NMPA and is applicable to PD-L1 tumor proportion score. (TPS) ≥1%First-line Treatment for EGFR Mutation-negative and ALK-negative Metastatic Non-small Cell Lung Cancer, becomingChina's First Approved Dual Immunotherapy Combination for Lung Cancer。

This approval is based on the CheckMate-227 study, the world's first Phase III clinical trial investigating a dual immunotherapy regimen for first-line treatment of non-small cell lung cancer (NSCLC), which evaluated the efficacy of Opdivo in combination with Yervoy versus chemotherapy. It is also one of the Phase III studies with the longest follow-up time in the field of NSCLC immunotherapy to date. Data confirms that this regimen provides long-term, sustained survival benefits.Enabling 22% of PD-L1≥1% patients to achieve a 6-year long-term survival(Chemotherapy group 13%), while avoiding chemotherapy toxicity, offering a new option for high-quality long-term survival for this patient population.
The primary endpoint results of the study showed that in patients with PD-L1≥1%, Opdivo combined with YervoyCan significantly improve median overall survival (mOS) Up to 17.1 months, 14.9 months in the chemotherapy group, hazard ratio [HR]=0.79, P=0.007.
Opdivo Combined with Yervoy GroupMedian Duration of Response (mDoR) Over 2 years, reaching 24.5 months,(Chemotherapy group was only 6.7 months), and 66% of the 5-year survival patients did not receive subsequent systemic treatment.(Only 20% in the chemotherapy group), indicating that this regimen can provide long-term stable survival benefits.
In terms of safety, the study showed that the safety of Opdivo (O药) combined with Yervoy (逸沃) is controllable and manageable: adverse events related to treatment of any grade and grades 3-4.Case (TRAE)The incidence rates were 77% and 33%, respectively, while in the chemotherapy group, they were 82% and 36%, respectively.
Exploratory analysis of the study further showed that in populations with mutations such as KEAP1, STK11, or TP53, who previously benefited less from immunotherapy combined with chemotherapy, the combination of Opdivo and Yervoy reduced the risk of death by 69%, 22%, and 28% respectively compared to chemotherapy alone. This suggests consistent benefits of this regimen for the aforementioned populations.
In addition, in the Chinese bridging trial CheckMate-227 CHESS, a consistent trend of benefit was observed in the Chinese population as seen globally.
On July 28, Antengene Corporation announced,Selinexor Tablets(Product Name:Xevio®)The new indication has been officially approved by the NMPA.In combination with bortezomib and dexamethasone(XVd Regimen), applicable to multiple myeloma patients who have previously received at least one line of treatment.(MM)Adult Patients。
The XVd regimen showed better clinical efficacy compared to the Vd regimen in Chinese R/R MM patients, as observed.Longer PFS and DOR, achieving higher ORR, ≥VGPR ratio, and MRD negativity rate, and showing a trend of OS extension; Elderly patients benefit significantly:Efficacy in the subgroup of elderly patients aged ≥65 years was significant.,SelinexorProvides a better option for this group of people.
This is the third indication for Selinexor to be approved in China, with two previous indications already approved: 1)Monotherapy for Relapsed/Refractory Diffuse Large B-Cell Lymphoma(R/R DLBCL);2)In combination with dexamethasone for the treatment of R/R MM, all haveIncluded in the National Medical Insurance Catalog.
Selinexor is the world's first orally administered selective XPO1 inhibitor with a novel mechanism introduced by Antengene.2023In August, Antengene Corporation entered into a collaboration withHansoh PharmaSigning regardingCommercializing Selinexor and other products containing or composed of Selinexor in mainland ChinaCooperation Agreement. Antengene will receive up to RMB200 million yuanThe down payment, and up to RMBRMB 5.35 billionMilestone payment.
On July 28, the NMPA official website announced that Qilu Pharmaceutical had submitted a new drug application for category 2.2.Risperidone Oral Soluble FilmApproved for marketing. The indication approved this time isBipolar Disorder and Schizophrenia. According to the Insight database, this is also the first approved in ChinaRisperidone Oral Soluble Film.

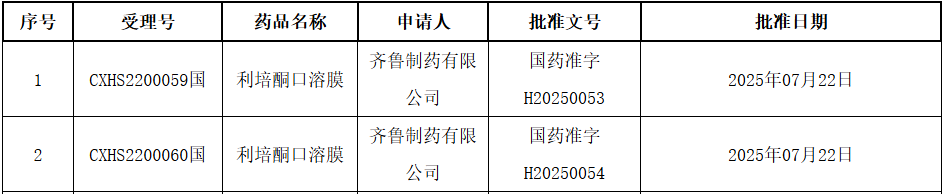
Risperidone is a second-generation antipsychotic drug commonly used in clinical practice. Its main mechanism of action is the antagonism of dopamine D2 receptors and 5-HT2A receptors, regulating the balance of central neurotransmitters. In 2013, Qilu Pharmaceutical initiated the development of risperidone oral soluble film. The oral soluble film formulation has several advantages, including:
Convenient to take, not easy to spit out, no risk of choking, the drug can quickly disintegrate upon contact with a small amount of saliva in the mouth, and does not require chewing or water for swallowing, greatly improving compliance among patients, especially the elderly, children, those with swallowing difficulties, bedridden patients, those lacking access to water outdoors, and patients who are not actively cooperative with medication.
Convenient to carry, compact in appearance, generally the size of a thin film chewing gum, with a good taste, and higher user acceptance.
Fast absorption, rapid onset of action.
According to the Insight database,As of now,Qilu Has Submitted Marketing Applications for 7 Oral Soluble Film Formulations in China, including brexpiprazole orally disintegrating film, risperidone orally disintegrating film, aripiprazole orally disintegrating film, olanzapine orally disintegrating film, memantine hydrochloride orally disintegrating film, montelukast sodium orally disintegrating film, tadalafil orally disintegrating film, etc. Except for brexpiprazole orally disintegrating film, all other products have been approved.


July 30, the NMPA official website shows,AstraZeneca Long-Acting C5 Complement Inhibitor "Relatlimab"Approved for a new indication in China(Application No.:JXSS2400086/7). According to the progress of relevant clinical trials, this indication is most likely to be:Used forAQP4 Antibody-Positive Neuromyelitis Optica(NMOSD)Adult patients.

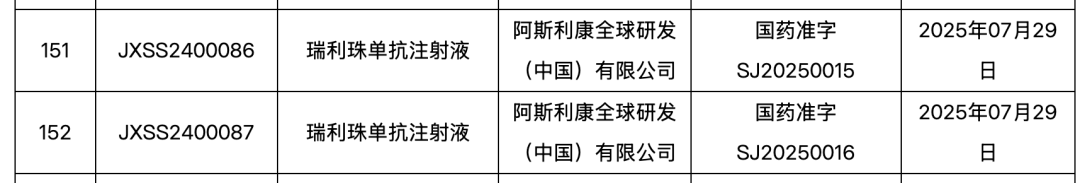
Relatlimab(English trade name: Ultomiris; Chinese trade name: 伟立瑞)It was AstraZeneca's investment of$39 billionImportant Rare Disease Pipeline Acquired After the Heavyweight Purchase of Alexion.On April 15, 2025, the drug was approved for marketing in China for the first time.,Combined with conventional therapeutic drugs for the treatment of anti-acetylcholine receptor(AChR)Adults with positive antibodiesGeneralized Myasthenia Gravis(gMG)Patient。The approval this time is for Relizumab's application in China.The Second Indication。
In China, the indications for gMG and NMOSD have been gradually approved.After the batch,Ravulizumab for the Phase III Clinical Trial in PNHIt has also been registered and announced in July 2024, with the first patient enrolled in October of the same year. By November, 18 patients had been recruited, and the trial is expected to be completed by January 2026. Subsequently, the commercialization process is expected to proceed steadily in China.



Screenshot from: NMPA official website
Sokazolimab(Socazolimab)It is a PD-L1 monoclonal antibody screened based on Sorrento's patented G-MAB library platform and was later introduced by Lee's Pharmaceutical. The drug initiated clinical trials for the first time in June 2018, filed for marketing authorization for the first time in October 2021, and was approved for marketing in December 2023.The first approved indication is cervical cancer.。
A total of 498 patients were enrolled in the trial and randomly assigned in a 1:1 ratio to receive sokazolimab+Carboplatin + EtoposideGroup and placebo+Carboplatin + EtoposideGroup. The research results show:
Socazolimab GroupmOS was 13.9 months(95%CI,12.22 - 15.34), while the placebo group was11.58 months(95%CI,10.64 - 12.81), HR is 0.799(95%CI,0.652-0.979,P = 0.0158)。 Socazolimab Group and Placebo GroupThe 1-year OS rates were 56.8%(95%CI,50.4-62.7)And48.8%(95%CI,42.4-54.8);2-Year OS RateRespectively 20.7%(95%CI,14.8-27.3)And5.9%(95%CI,0.8-18.9)。 Secondary endpointsSocazolimabTreatment group and placebo groupmPFS were 5.55 months and 4.37 months, respectively.,ORR were 75.5% and 68.1%, respectively.。
On July 31, AstraZeneca and MSD jointly announced that the PARP inhibitorOlaparib TabletsApproved for a new indication in China, in combination with abiraterone and prednisone or prednisolone forCarrying germline or somatic BRCA mutations(gBRCAm or sBRCAm)Metastatic Castration-Resistant Prostate Cancer(mCRPC)Treatment of adult patients.

Screenshot source: Official WeChat account of the company
The approval of this new indication is based on the subgroup analysis results from the global cohort and the China cohort of the PROpel Phase III trial. Data from BRCA-mutated patients in the global cohort showed that, compared to abiraterone monotherapy, the combination of olaparib and abiraterone demonstrated improvement in radiographic progression-free survival. (rPFS) ...showed clinically significant improvements. Although the sample size of the Chinese cohort was relatively small, which limited the interpretation to some extent, the study data still demonstrated improvements consistent with the trend observed in the global cohort.
National TeamPatients with BRCA Mutation SubgroupThe analysis results show that,Olaparib Combined with Abiraterone Reduces Risk of Disease Progression or Death by 76% and Risk of Death by 70%. The median rPFS and median overall survival in the olaparib plus abiraterone group(OS)Not yet reached; the median rPFS and median OS in the abiraterone monotherapy group were 8 months and 23 months, respectively.
In the global cohort of the PROpel III phase trial, the safety and tolerability of olaparib combined with abiraterone were consistent with the known safety profiles from previous clinical trials and each monotherapy.
In the Chinese cohort, patients with BRCA mutations showed a consistent trend in efficacy as observed in the global cohort with BRCA mutations. The safety results in the Chinese cohort were consistent with those in the global cohort, with no new safety issues identified.
Olaparib is the world's first PARP inhibitor. To date, olaparib has been approved for 7 indications in China.
On July 28, Akeso Biopharma announced that the third indication for its Ivonescimab Injection has been accepted for marketing application, with the indication beingFirst-line Combination Chemotherapy for Advanced Squamous Non-Small Cell Lung Cancer(sq-NSCLC)。

Source of Screenshot:AkesoOfficial WeChat
Ivonescimab, a globally first-in-class PD-1/VEGF bispecific antibody independently developed by Akeso Biopharma, was approved for marketing in China for the first time in May 2024. It is indicated for locally advanced or metastatic nsq-NSCLC with disease progression after EGFR-TKI treatment. In April this year, the product received regulatory approval for a new indication as a first-line treatment for advanced NSCLC with positive PD-L1 expression.
The current applicationThe new first-line treatment indication for sq-NSCLC will drive Iovocin to achieve a more comprehensive layout in the NSCLC field, covering both first-line and later-line treatments, as well as squamous and non-squamous patient populations.。
The sNDA for the new indication is based on the Phase III clinical trial.(AK112-306/HARMONi-6 Study)Positive results. The HARMONi-6 study aims to evaluateEvolisib Combined with Chemotherapy "Head-to-Head" Tislelizumab Combined with Chemotherapyeffect. In April this year, Akeso Biopharma announced that the Independent Data Monitoring Committee(IDMC)The pre-specified interim analysis of the evaluation showed strongly positive results:
In the intent-to-treat population (ITT) In China,Progression-Free Survival in Patients Treated with Yivoxi Combined with Chemotherapy(PFS)Compared with the control group, a decisively positive result was obtained.;
Compared with the control group, the Yivoxi group showed significant PFS benefits with clinical significance in both PD-L1 positive and PD-L1 negative populations.
This clinical trial enrolled a total of 532 subjects, with centrally located squamous cell carcinoma accounting for approximately 63%, consistent with the distribution of patients in the real world.
The overall safety of the Yivoxi group was good, with no new safety signals identified. The incidence of treatment-related serious adverse reactions and the incidence of grade 3 or higher bleeding events were similar to those in the control group.
July 30, the CDE official website shows,Amgen andBeiGene Jointly Submitted InjectableTalazoparibNew indications for marketing application have been accepted. Based on the clinical trial progress of this drug, the indications applied for in this submission are:Used forSecond-line Treatment for Small Cell Lung Cancer Patients。
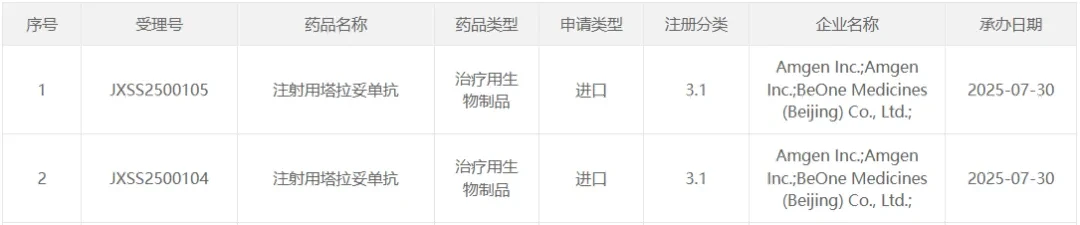
Source of screenshot:CDE Official Website
Talatotumab is a product developed byAmgenThe innovative targeted immunotherapy developed by the company can simultaneously bind to the DLL3 protein on tumor cells and the CD3 protein on T cells, thereby activating T cells to kill tumor cells expressing the DLL3 protein. In October 2019, Amgen and BeiGene reachedStrategic Cooperation,This includes jointly advancing the development and commercialization of Taladotinib in China.
Based on the progress of the clinical trials of this drug, the indication for this application is speculated to be:Used forPatients with recurrent small cell lung cancer after receiving platinum-based first-line chemotherapy。
In the randomized, open-label international multicenter (including China) Phase III study DeLLphi-304, researchers compared the efficacy and safety of talactofugine with standard therapy in subjects with recurrent small cell lung cancer after receiving platinum-based first-line chemotherapy.
In April 2025, Amgen announced that the DeLLphi-304 study had met its primary endpoint in a planned interim analysis. Compared with local standard treatments, (SOC) Compared with chemotherapy, taladotinMedian Overall Survival (mOS) For 13.6 months(vs 8.3 months), demonstrating statistically significant and clinically meaningfulImprovement. The safety of Taladotinib is consistent with its known characteristics.(See the figure below for details)。
On July 31, the CDE website showed that Huadong Medicine's Edaravone Tablets(Research and Development Code: TTYP01)The listing application has been accepted, with the indication being:Improve Acute Ischemic Stroke(AIS)Neurological symptoms and functional disorders caused, improve daily living abilities。

Source: CDE Official Website
Edaravone is a novel free radical scavenger approved in Japan and China for the treatment of AIS. Edaravone injection requires intravenous administration, which limits patient compliance and convenience with long-term use. Currently, all approved products have a treatment duration of 14 days.
TTYP01 TabletsDeveloped independently by Auson BiotechEdaravone Oral Tablets, an improved new drug that has not been marketed both in and outside of China. On July 12, 2024, Sinopharm Huadong and Auson Biotech reached an exclusive licensing agreement with a total amount exceeding 1.285 billion yuan,Obtained TTYP01 Tablets from Auson Biotech(Edaravone Tablets)Exclusive license for all indications in mainland China, Hong Kong, Macao, and Taiwan, including development, registration, production, and commercialization rights.
TTYP01 tablets do not rely on professional medical personnel for injection; patients can self-administer the drug. The tablets can be transported and stored at room temperature, offering greater convenience and compliance. Data from the Phase III clinical study completed in June 2024 showed that, under the premise that the efficacy and safety of edaravone tablets are not inferior to the improved drug edaravone injection, compliance was improved, supporting its application for market approval.
July 29Recently, the CDE official website showed that Chia Tai TianqingTQC3721 Inhalation Suspension Proposed for Inclusion in Breakthrough Therapy Designation, useInChronic Obstructive Pulmonary Disease(COPD)Maintenance Treatment。
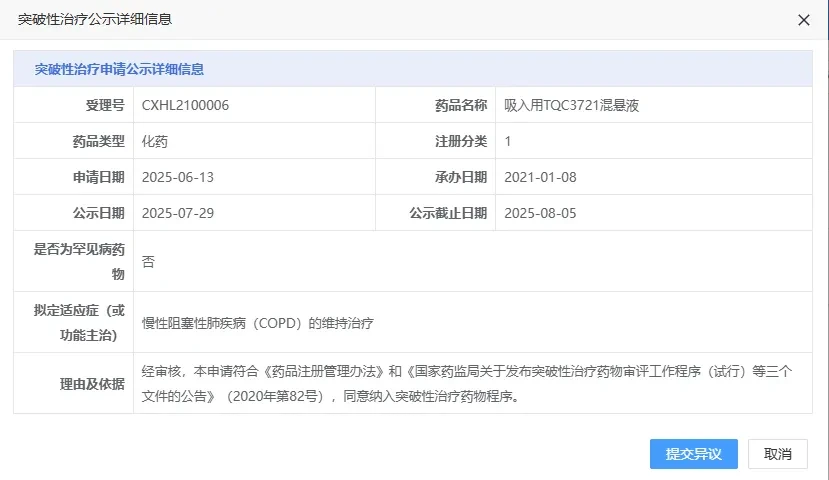
Screenshot source: CDE official website
TQC3721 is aA Novel Inhaled PDE3/4 Inhibitor with a New Mechanism, with dual effects of bronchodilation and anti-inflammation, thereby alleviating patient symptoms, suppressing inflammation, and controlling disease progression.
Clinical study results show that TQC3721 in COPD patients with single-bronchodilator and dual-bronchodilator background therapy,Significant improvements in lung function and symptom scores, and the relevant data will be announced at an upcoming international academic conference.
It is worth mentioning that,The inhalation suspension TQC3721 has been approved by the CDE.Approval for ImplementationPhase IIIRegistered clinical study for the maintenance treatment of COPD. Compared with the marketed PDE3/4 products, the Phase III clinical study of TQC3721Will additionally include patients on dual bronchodilator background therapy, covering a broader population of COPD patients.
In addition, except for inhalation suspensions,TQC3721 Inhalation PowderCurrently in Phase I clinical development, the dry powder formulation is expected to further improve patient compliance.
On July 30, the official website of the Drug Clinical Trial Registration and Information Disclosure Platform showed that HengruiA pharmaceutical registration was filed for one item.Evaluation of SHR-1905 Injection in Patients with Severe Uncontrolled AsthmaEfficacy and Safety of ParticipantsPhase III Clinical Study of Sexuality(SHR-1905-302, Registration No.:CTR20252988)。
This is aA multicenter, randomized, double-blind, placebo-controlled, parallel-design Phase III clinical trial, planning to enroll 408 subjects, who will be randomly assigned to receiveSHR-1905 Injection, Placebo Treatment.The principal investigator isZhong Nanshan, Chief Physician of the First Affiliated Hospital of Guangzhou Medical UniversityAnd Deputy Chief PhysicianYang Xinyan.
The primary endpoint isAnnual Acute Attack Rate of Asthma(AAER), secondary endpoints includeWeek 48, Before Using Bronchodilator(Pre-BD)Forced Expiratory Volume in One Second(FEV1)Change from Baseline,Exhaled Nitric Oxide(FeNO)Changes from baseline, etc.
SHR-1905 is a thymic stromal lymphopoietin independently developed by Hengrui.(TSLP)Monoclonal antibodies can block the release of inflammatory cytokines, inhibit downstream inflammatory signaling, and ultimately improve the inflammatory state and control disease progression.
In August 2023, Hengrui and U.S.-based One Bio(Later calledAiolos Bio)Reach an agreement to grant a paid license for the SHR-1905 injection project to the latter,Upfront and near-term milestone payments of $25 million, with R&D and sales milestone payments not exceeding $1.025 billion. In early 2024, GSK acquired Aiolos Bio for $1.4 billion, obtaining the product.
2. Zhongsheng Pharmaceutical: Initiation of Phase III Clinical Trial for GLP-1R/GIPR Agonist, Head-to-Head with Semaglutide
On July 30, the official website of the Drug Clinical Trial Registration and Information Disclosure Platform showed that Zhenzhong Pharmaceutical, a subsidiary of Zhenzhong PharmaceuticalRuichuang registered aRAY1225 Injection Combined Therapy for Type 2 Diabetes Phase III Clinical Trial(RAY1225-24-13, Registration No.:CTR20252996)。
This is aA multicenter, randomized, open-label, Phase III clinical trial with semaglutide injection as control, aimed atEvaluate the safety and efficacy of RAY1225 in patients with type 2 diabetes whose blood glucose levels are poorly controlled after treatment with oral hypoglycemic agents.
The study plans to enroll600 subjects, randomly assigned to receiveRAY1225 is administered once every two weeks by subcutaneous injection.AndSemaglutide is administered once weekly by subcutaneous injection.Treatment, lasting 52 weeks. The primary endpoint isChange in HbA1c from baseline at 36 weeks of treatment, secondary endpoints includeChanges in HbA1c from baseline at each time point,Changes in HbA1c from baseline at each time point,Adverse events, laboratory tests, vital signs, electrocardiogram, hypoglycemic events, etc.
RAY1225 is a self-developed product by Zhongsheng Ruichuang.GLP-1/GIP Dual Receptor AgonistRAY1225 promotes insulin secretion in a glucose-dependent manner, delays gastric emptying, and regulates human metabolism through multiple mechanisms, thereby achieving hypoglycemic, weight-reducing, and peripheral-lowering effects.Insulin ResistanceAnd other effects, it is expected to be used in the treatment of various metabolic diseases such as hypoglycemia, weight loss, and metabolic syndrome.
July 28,Drug Clinical Trial Registry and Information Disclosure Platform Shows, BeiGene/Amgen registered aTarlatamab(Talazoparib)Used forUntreated extensive-stage small cell lung cancer(ES-SCLC)Phase III clinical trial of the patient.
This is aPhase III, Open-label, Multicenter, Randomized Study(DeLLphi-312), aiming toComparisonTalazoparib, Durvalumab, Carboplatin, and Etoposide versus Durvalumab, Carboplatin, and Etoposide as First-Line Treatment for ES-SCLC: Efficacy, Safety, and Tolerability.
The study plans to enroll 60 participants in China and 330 participants internationally.The primary endpoint of the study isOverall Survival(OS), secondary endpoints includeProgression-Free Survival(PFS), Objective Response(OR), Disease Control(DC)And the duration of relief(DOR)and other aspects.
Talatodermab is a product developed byAmgenThe innovative targeted immunotherapy developed by the company can simultaneously bind to the DLL3 protein on tumor cells and the CD3 protein on T cells, thereby activating T cells to kill tumor cells expressing the DLL3 protein.
In October 2019, Amgen and BeiGene reachedStrategic Cooperation,This includes jointly advancing the development and commercialization of Taladotinib in China.
From the ongoing Phase III clinical trials, Amgen and BeiGene have already initiated studies on this drug.Research on Third-line, Second-line to First-line in Small Cell Lung Cancer, also covering Maintenance/Consolidation Therapy. Among them, the third-line therapy has been approved in China recently.Report on Market LaunchAnd has entered the priority review channel.


