Abbott Receives FDA 510(k) Clearance for Alinity m EBV Assay with Alternative DNA Polymerase

Abbott Molecular
Molecular Diagnostics Researcher

Abbott
Diagnostic and pharmaceutical product manufacturers

In the postoperative management of organ transplant patients, monitoring for Epstein-Barr Virus (EBV) is crucial—it not only can lead to fatal post-transplant lymphoproliferative disorder (PTLD) but also directly influences adjustments to the immunosuppressive regimen.
Recently, Abbott Molecular, a molecular diagnostics company under Abbott, announced that its next-generation fully automated nucleic acid testing system, Alinity m EBV, has received FDA 510(k) clearance in the United States. It is used for quantitatively detecting EBV DNA in the plasma of transplant patients, enabling more precise clinical management of infection risks.
Notably, this new reagent is not an "entirely new species" but a minor iteration of Abbott's already marketed product, Alinity m EBV (K212778, approved in 2022) — the only change being that another DNA polymerase, MomentaTaq, can now be used in the reagent formulation as an alternative to the original KAPA2G. This modification has not introduced any new safety or efficacy concerns.
So, why is this "fine-tuned" reagent still worth attention? What substantial improvements does it bring to transplant patient management?
In today's article, I will break down its technical details and clinical value for everyone based on the full text of the product evaluation report.

Product Overview
Alinity m EBV is an in vitro polymerase chain reaction (PCR) testing device used for the quantitative detection of Epstein-Barr virus (EBV) DNA in human plasma.
This device is essentially consistent with the previously approved similar product (Registration No. K212778) in overall design and operational procedures. The only difference is that this device allows the use of MomentaTaq DNA polymerase as a substitute for KAPA2G DNA polymerase in the reagent formulation, but this substitution does not impact sample processing, testing steps, or data analysis.
To support the formulation using MomentaTaq DNA polymerase, additional research was conducted, and these supplementary data were combined with the previously submitted analytical and clinical testing data for K212778.
The entire Alinity m EBV testing process includes sample preparation, real-time PCR assembly, amplification/detection, result calculation, and reporting, all of which are fully automated by the Alinity m system without any intermediate handling or transfer operations required by the user.
This system belongs to the random access analyzer and can run in parallel with other Alinity m assays on the same instrument.
This test requires three independent specific kits:
First is the Alinity m EBV AMP Kit, which includes two types of multi-well reagent trays – the amplification tray (AMP TRAY 1) contains lyophilized, single-dose PCR amplification/detection reagents and lyophilized, single-dose internal control (IC), each placed in separate wells; the activation tray (ACT TRAY 2) contains liquid, single-dose activation reagents.
The kit is recommended to be stored at 2°C to 8°C.
Next is the Alinity m EBV CTRL kit, which includes a negative control, a low positive control, and a high positive control, all in liquid form, contained in single-use tubes, with recommended storage at -25°C to -15°C.
Finally, the Alinity m EBV CAL Kit, which includes calibrators at two concentration levels, is also in liquid form, packaged in single-use tubes, with a recommended storage condition of -25°C to -15°C.
EBV DNA in human plasma is automatically extracted on the Alinity m system using the built-in Alinity m Sample Preparation Kit 2, Alinity m Lysis Buffer, and Alinity m Diluent.
The system uses magnetic microparticle technology to achieve the capture, washing, and elution of nucleic acids.
The purified nucleic acid is then mixed with a liquid single-dose activation reagent and a lyophilized single-dose amplification/detection reagent, and transferred to a reaction vessel.
After adding the Alinity m vapor barrier solution, the reaction vessel is transferred to the amplification/detection unit for PCR amplification and real-time fluorescence detection of the EBV target.
During the initial stage of sample preparation, the lyophilized internal control on the AMP disc is reconstituted by the system and added to the reaction of each sample. This internal control undergoes the complete sample preparation and PCR process together with the samples, calibrators, and controls to verify whether the sample handling is appropriate and the test results are valid.
The amplification/detection reagent contains the enzymes, primers, probes, and activation reagents necessary for polymerization and detection.
To determine the concentration of EBV DNA in the sample, an EBV calibration curve must be established. This is achieved by processing calibration samples at two concentration levels and performing sample preparation and PCR. Subsequently, the EBV DNA concentration in the samples and controls is calculated based on the stored calibration curve.
To ensure the continuous reliability of instrument and reagent performance, the detection process will test the control samples at a predetermined minimum frequency. Each control event includes a negative control, a low positive control, and a high positive control, which undergo the same sample preparation and PCR procedures as the samples.
In addition, the test also relies on a series of other components and software, including Alinity m EBV application specification documents, the Alinity m system and its system software, Alinity m Sample Preparation Kit 2, Alinity m Sample Dilution Kit I, Alinity m system solutions (such as lysis solution, diluent, vapor barrier solution), as well as various Alinity m dedicated tubes and caps, etc.

Intended Use
Alinity m EBV is an in vitro polymerase chain reaction (PCR) assay for the quantitative determination of Epstein-Barr virus (EBV) DNA in human EDTA plasma on the automated Alinity m system.
Alinity m EBV is designed to assist in managing EBV in transplant patients.
In patients monitored for EBV, sequential DNA measurements may be used to indicate the need for potential treatment modifications and to assess viral response to therapy.
The results of Alinity m EBV must be interpreted in the context of all relevant clinical and laboratory findings.
Alinity m EBV should not be used for screening blood, blood products, or human cells, tissues, and cellular- and tissue-based products (HCT/Ps) donors for EBV detection.

Comparison with the product before the change
The Alinity m EBV test reagent is substantially equivalent to the currently marketed product (Registration No. K212778) in terms of its main functional components. Both belong to legally marketed nucleic acid amplification tests (NAAT) used for the quantitative detection of Epstein-Barr virus (EBV) DNA.
These two reagents have exactly the same intended use, both designed for the quantitative detection of EBV DNA in EDTA-anticoagulated human plasma on the automated Alinity m system, to assist in the management of EBV in transplant patients.
Continuous monitoring of changes in EBV DNA levels in patients can indicate whether treatment adjustments are necessary and assess the response of the virus to the treatment, but the test results must be interpreted in conjunction with all relevant clinical and laboratory information.
In addition, this assay is not intended for use as an EBV screening test for blood donors, blood products, or HCT/Ps donors (human cells, tissues, and cellular and tissue-based products).
The two reagents are consistent in several key aspects: both are used for quantitative detection, targeting two highly conserved regions (gp350 and EBNA1) of the EBV genome. The sample types processed are EDTA anticoagulated plasma, and the sample preparation process employs automated liquid handling and robotic operation platforms. The amplification technology is based on real-time polymerase chain reaction (PCR), and both use the same detection controls, including negative control, low positive control, high positive control, and internal control (IC).
These commonalities ensure a high degree of similarity between the two reagents in terms of detection principles, procedures, and result interpretation.
The main difference between the two reagents lies in the different DNA polymerases used.
The marketed product only uses KAPA2G HotStart DNA Polymerase for DNA amplification, while the currently applied Alinity m EBV reagent allows the use of either KAPA2G or MomentaTaq HotStart DNA Polymerase.
This difference is limited to the type of polymerase used in the reagent formulation and does not introduce any new safety or efficacy concerns. It also does not impact the operational procedures, sample handling methods, or result analysis of the test.

Limit of Detection
To support the previously approved registration number K212778, a study on the Limit of Detection (LoD) was conducted, and relevant content can be found in the review report for that registration number.
An additional study was conducted for the Alinity m EBV reagent (using the MomentaTaq DNA polymerase formulation) in this application to confirm its claimed limit of detection (20 IU/mL).
This study evaluated the limit of detection for EBV type 1 by testing dilutions of the first edition of the World Health Organization (WHO) International Standard for Epstein-Barr Virus (EBV) Nucleic Acid Amplification Techniques (NIBSC code 09/260) added to EBV-negative human plasma.
Each concentration of EBV DNA was tested multiple times over several days using three different batches of amplification reagents.
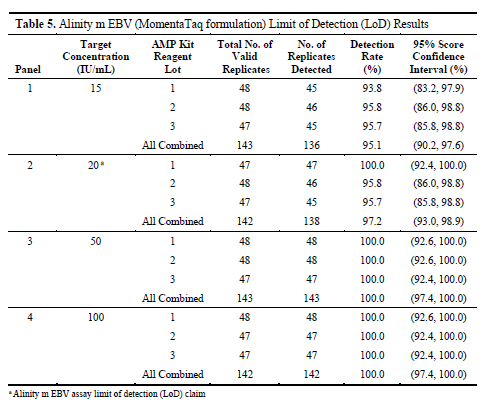
Research data shows that the overall detection rate of Alinity m EBV reagent formulated with MomentaTaq DNA polymerase at a concentration of 20 IU/mL, integrating results from three reagent batches, is 97.2%, with a 95% confidence interval of (93.0%, 98.9%).
This means that at this concentration, the vast majority of samples can be reliably detected, and the results have a high degree of credibility.
The study also tested lower (e.g., 15 IU/mL) and higher (e.g., above 20 IU/mL, including 50 IU/mL and 100 IU/mL) concentration levels respectively.
At 15 IU/mL, the overall detection rate was 95.1%, while at concentrations of 20 IU/mL and above, the detection rate reached or approached 100%.
Particularly at the critical concentration of 20 IU/mL, out of a total of 142 valid replicate samples across three reagent batches, 138 were successfully detected, achieving a detection rate as high as 97.2%, thus supporting the designation of this concentration as the limit of detection for the reagent.
In contrast, at concentrations of 50 IU/mL and 100 IU/mL, all samples were detected 100%, further verifying the sensitivity and reliability of the reagent in higher concentration ranges.

Limit of Quantitation
To verify the claimed lowest limit of quantification (LLoQ, 50 IU/mL) of the Alinity m EBV assay reagent formulated with MomentaTaq DNA polymerase, a specific study was conducted.
Previously, relevant research was conducted to support the approval of registration number K212778. For more details, please refer to the decision summary for this registration number.
The lowest limit of quantitation refers to the minimum concentration at which the detection system can reliably quantify the target substance (here, EBV DNA) without exceeding the acceptable total error range.
In other words, the LLoQ not only requires the detection system to detect the presence of this concentration, but also demands that it provides quantitative results close to the true value with sufficient accuracy and precision.
In this study, the researchers analyzed a series of samples with known concentrations, which were prepared by diluting the first edition of the World Health Organization International Standard for EBV Nucleic Acid Amplification Techniques (NIBSC code 09/260) and adding it to EBV-negative plasma.
The study focused on multiple low-concentration samples, including 50 IU/mL (i.e., 1.70 Log IU/mL), to evaluate whether the accuracy and precision of the test results at these low concentrations met the established standards.
To determine whether 50 IU/mL can indeed be used as the lowest limit of quantitation, the study employed two methods to estimate total error: one is Total Analytical Error (TAE), calculated as bias + 2 × standard deviation (SD); the other is Total Error (TE), calculated as √2 × 2 × standard deviation.
These two methods evaluate the possible range of measurement deviation from the true value from different perspectives, thereby helping to determine whether the test results are sufficiently reliable at this concentration.
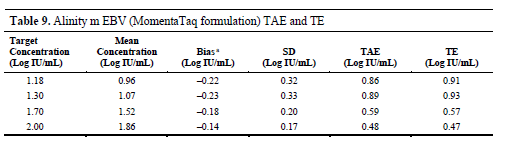
The research results show that at the concentration of 50 IU/mL (1.70 Log IU/mL), the deviation of the test results is small, and the standard deviation is also at a low level. The calculated total analytical error (TAE) is 0.59 to 0.86 Log IU/mL, and the total error (TE) is 0.47 to 0.91 Log IU/mL, both of which are less than or equal to the acceptable standard of 1.00 Log IU/mL.
This means that, at this concentration, the detection system is not only able to stably detect EBV DNA, but also the deviation and fluctuation between its quantitative results and the true value are within the clinically and laboratory acceptable range, providing a reliable basis for clinical decision-making.
In addition, the study also analyzed samples with lower concentrations (such as 1.18 Log IU/mL and 1.30 Log IU/mL, corresponding to approximately 15 IU/mL and 20 IU/mL), and found that although signals could be detected at these lower concentrations, the total error exceeded the acceptable range, indicating that the reliability of quantitative results cannot be guaranteed at these levels.
Therefore, considering all the indicators, it is reasonable and scientific to finally confirm 50 IU/mL (1.70 Log IU/mL) as the lowest limit of quantitation for this reagent.

Linear Range
To further confirm the linear performance of the Alinity m EBV assay reagent, formulated with MomentaTaq DNA polymerase, within its claimed quantitative range, an additional linearity study was conducted.
Previously, a linearity study was also conducted to support the approval of registration number K212778. For more details, please refer to the decision summary of this registration number.
The purpose of this study is to verify whether the reagent can maintain a good linear relationship within the range from 50 IU/mL (i.e., 1.70 Log IU/mL) to 200,000,000 IU/mL (i.e., 8.30 Log IU/mL), meaning whether the test results can proportionally and accurately reflect the EBV DNA concentration in the actual samples across this concentration span.
To evaluate linearity, the researchers used a set of 16 samples with different concentration levels. The target EBV DNA concentrations of these samples covered the entire expected quantitative range, and also intentionally included samples with a concentration of 15 IU/mL, which is below the expected lower limit of quantitation (LLoQ), as well as samples with a concentration of 250 million IU/mL, which is above the expected upper limit of quantitation (ULoQ), in order to more comprehensively examine the performance of the reagent under extreme concentrations.
These sample discs contain samples prepared from different sample types (including plasmid DNA, cultured viruses, and clinical samples) and are designed with at least 2 log units of overlap between adjacent concentrations to ensure a reasonable and continuous concentration gradient, facilitating accurate analysis of linear trends.
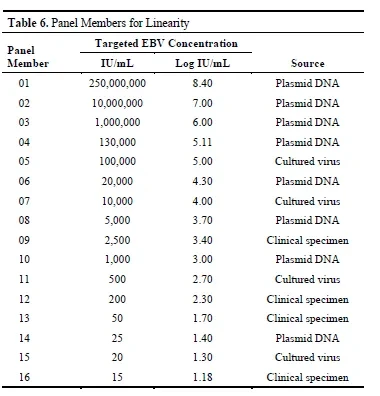
The study results show that the Alinity m EBV reagent using MomentaTaq DNA polymerase demonstrates good linearity across a broader concentration range of 15 IU/mL to 250 million IU/mL (i.e., 1.18 Log IU/mL to 8.40 Log IU/mL).
In other words, within this range, the detection system can accurately reflect the true concentration of EBV DNA in the sample, and there is a reliable proportional relationship between the test results and the actual concentration.
Although the official claimed quantitative range of the reagent is 50 IU/mL to 200 million IU/mL, actual tests show that it can maintain a linear response at both lower and higher concentrations, further demonstrating the stability and accuracy of the detection method across a broader concentration range.
These data indicate that the reagent can not only provide accurate quantitative results within the conventional clinically relevant concentration range, but also exhibits reliable linearity at extremely low and high concentrations, offering broader application possibilities for testing needs in various clinical scenarios.
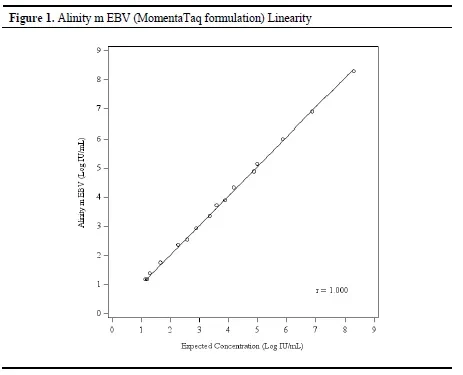
The overall linear performance is visually reflected in this consistency through graphical representation (as shown in Figure 1), demonstrating a good correspondence between the detection signal and the actual concentration.

Precision
To verify the performance of the Alinity m EBV test reagent formulated with MomentaTaq DNA polymerase in terms of precision and clinical reproducibility, additional studies were conducted to confirm whether it meets the established performance claims and to compare it with similar reagents previously using KAPA2G DNA polymerase.
These studies are based on previous work conducted to support registration number K212778 and introduce new experimental data and analysis.
In the precision study, the goal is to confirm the consistency and stability of the reagent's test results when detecting EBV DNA across different concentration ranges.
Specifically, the study validated two key metrics: for plasma samples with concentrations ranging from 500 IU/mL to 200 million IU/mL (i.e., 2.70 Log IU/mL to 8.30 Log IU/mL), the intra-laboratory standard deviation (SD) must not exceed 0.25 Log IU/mL; whereas for samples with concentrations between 20 IU/mL and 500 IU/mL (i.e., 1.30 Log IU/mL to 2.70 Log IU/mL), the intra-laboratory SD must not exceed 0.50 Log IU/mL.
The study was evaluated using sample groups with eight different concentration levels. The EBV DNA concentrations of these samples ranged from 20 IU/mL to 200 million IU/mL and were prepared by dilution in human plasma using EBV-positive clinical samples, cultured virus, or synthetic DNA (for high-concentration samples only). All concentrations were traceable to the first edition of the World Health Organization International Standard for EBV (NIBSC code 09/260).
The experiment was conducted by three operators on the same Alinity m system using three different batches of MomentaTaq reagents. Each sample was tested three times per day for 15 consecutive days, with an interval of at least two hours between adjacent tests to adequately simulate the sources of variation in actual testing.
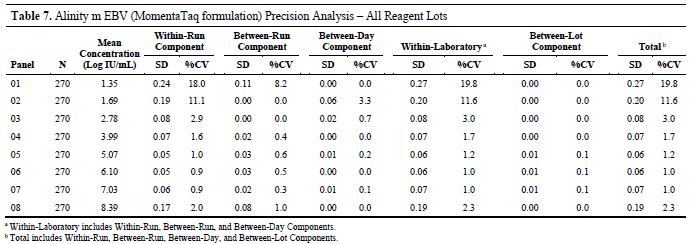
The study results showed that for samples in the higher concentration range (500 IU/mL to 200 million IU/mL), the intra-laboratory standard deviation was controlled at 0.25 Log IU/mL or lower; whereas for samples in the lower concentration range (20 IU/mL to 500 IU/mL), the standard deviation was controlled at 0.50 Log IU/mL or lower, meeting the pre-established precision requirements.
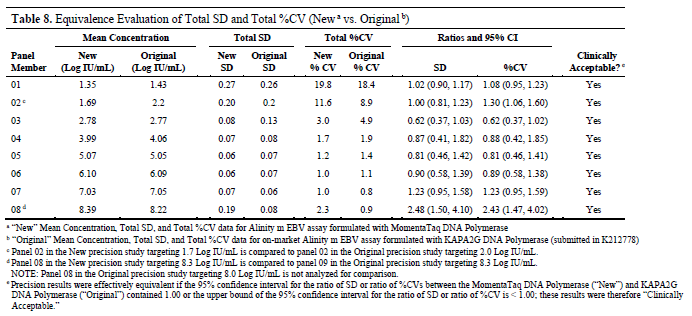
Further comparison of the total standard deviation and total coefficient of variation (%CV) between this study and the data submitted previously for K212778 revealed that the 95% confidence intervals of the ratios all contained 1.00 or had upper limits less than 1.00, indicating that the reagents using MomentaTaq demonstrated clinically acceptable precision consistency compared to the reagents using KAPA2G in the past.
In addition, a clinical reproducibility study was conducted to evaluate the performance consistency of the reagent across different laboratories, different operators, and different testing days.
The study selected 9 samples (8 positive samples and 1 negative sample), with the positive samples covering different concentration levels and spanning the entire quantitative range. The samples were prepared using EBV-positive clinical samples, cultured virus, or plasmid DNA in EDTA plasma.
The experiment was conducted at three different clinical centers, running twice daily for five consecutive days, with each sample being tested in four replicates per run to ensure at least three valid results were available for analysis.
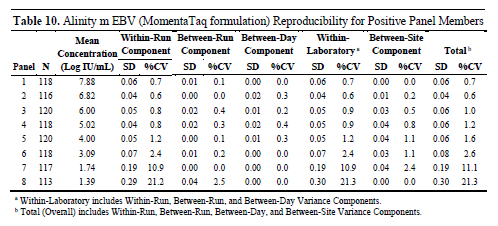
The results showed that among all positive samples, the internal precision of the laboratory was good, with both the standard deviation and coefficient of variation maintained at low levels, indicating a high degree of consistency in test results under different conditions.
Particularly in medium to high concentration samples, the variation is minimal, demonstrating excellent reproducibility; whereas in low concentration samples, although the variation is slightly higher, it remains within an acceptable range.
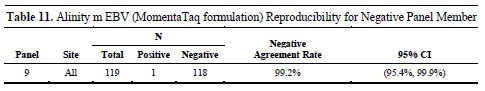
For the only negative sample, there was only one false positive result in all tests, with a negative agreement rate of 99.2%, further verifying the reliability of the reagent in actual testing.
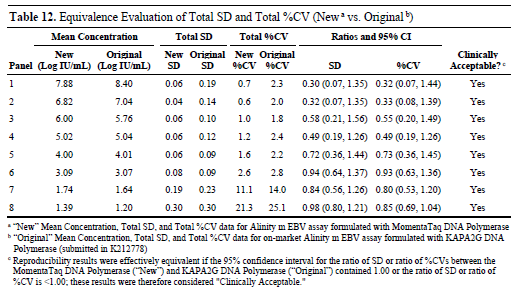
Compared with the original reproducibility study data, it was also found that the total standard deviation and total coefficient of variation in this study showed little difference from the previous data, and the 95% confidence interval of their ratio also met the clinically acceptable criteria.

Clinical Evaluation
To evaluate the performance of the Alinity m EBV assay (hereinafter referred to as "Alinity m EBV IUO") formulated with MomentaTaq DNA polymerase in actual clinical applications, particularly its consistency in test results with a commercially available assay formulated with KAPA2G DNA polymerase (i.e., the control assay, registration number K212778), a method comparison study was conducted.
The purpose of this study is to demonstrate the equivalence of these two reagents in detecting Epstein-Barr virus (EBV) DNA in human plasma, meaning they can provide similar and reliable quantitative results in practical use.
The study included a total of 124 samples, with the EBV DNA concentrations of these samples all falling within the common quantitative range of the two reagents, ensuring the validity and scientific rigor of the comparison.
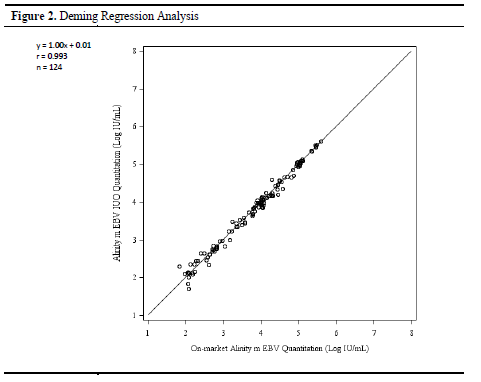
The detection results of these 124 samples were compared through Deming regression analysis. The results showed a very strong consistency between the two reagents: the slope of the regression line was 1.00, the intercept was 0.01, and the correlation coefficient reached 0.993.
These values indicate that the test results of Alinity m EBV IUO are highly linearly correlated with the control reagent, almost on the same straight line, showing that both perform very similarly when detecting samples of different concentrations.
From the bias analysis, the average bias of Alinity m EBV IUO's test results compared to the reference reagent is -0.01 Log IU/mL, with a 95% confidence interval of (-0.03, -0.01), indicating that overall, the quantitative results of Alinity m EBV IUO are slightly lower but this bias is very small, almost negligible, and is within an acceptable range both statistically and clinically.
The study further analyzed the systematic differences between the two reagents at four specific viral load levels (based on the detection results of the control reagent).
These four concentrations are the lowest limit of quantitation level (1.70 Log IU/mL, approximately 50 IU/mL), 3.00 Log IU/mL, 4.00 Log IU/mL, and 5.00 Log IU/mL.
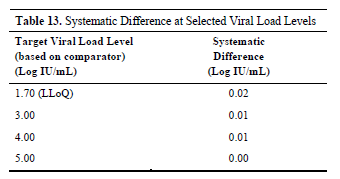
The results showed that the systematic differences between Alinity m EBV IUO and the reference reagent at these concentration points were very minimal, at 0.02 Log IU/mL, 0.01 Log IU/mL, 0.01 Log IU/mL, and 0.00 Log IU/mL, respectively.
This means that within these clinically significant concentration ranges, the test results of the two reagents are almost identical, whether at extremely low concentrations (close to the lower limit of quantitation) or at moderately high concentrations, with minimal differences in their quantitative values, further demonstrating their equivalence in actual clinical testing.

Three Key Questions
Alright, as before, I will use three key questions to tie together the relevant content of this review report.
The first question,What are the key steps in the sample processing of the Alinity m EBV device?
The sample processing procedure of the Alinity m EBV device includes the following key steps: First, the EB virus DNA in human plasma is automatically extracted using the Alinity m Sample Prep Kit 2, Alinity m Lysis Solution, and Alinity m Diluent Solution.
Then, nucleic acid capture, washing, and elution are performed using magnetic microparticle technology.
The purified nucleic acid is mixed with the liquid unit dose Alinity m EBV Activation Reagent and the lyophilized unit dose Alinity m EBV Amplification/Detection Reagent, and then transferred to the reaction vessel.
Finally, after adding Alinity m Vapor Barrier Solution, transfer the reaction vessel to the amplification/detection unit for PCR amplification and real-time fluorescence detection.
The second issue,How are the calibrators for the Alinity m EBV device used?
The Alinity m EBV instrument is calibrated using two levels of calibrators: Level 1 Calibrator and Level 2 Calibrator.
These calibrators are processed during sample preparation and PCR to generate a calibration curve.
Calculate the EB virus DNA concentration in the sample and control through the stored calibration curve.
The use of calibrators ensures the accuracy and reliability of test results.
The third question,What are the specific clinical application cases of the Alinity m EBV device?
The Alinity m EBV device has a wide range of clinical applications in the management of transplant patients.
For example, in patients after kidney transplantation, regular monitoring of EBV viral load using the Alinity m EBV assay can help doctors promptly detect signs of viral activity and adjust immunosuppressive treatment regimens. Q
In addition, the device can also be used for EB virus monitoring in patients undergoing liver transplantation, heart transplantation, etc., to evaluate treatment efficacy and patient prognosis.

Follow 【Diagnostic Science】, for more IVD R&D and registration insights!