Who Is the 'Best-in-Class' for DNA Methylation Profiling? A Comprehensive Comparison and Empirical Summary of Four Leading Technologies (2023–2025)

Illumina
Diagnostic Product Developer

DNA Methylation as a Core Mechanism of Epigenetic Regulation Plays a Crucial Role in Regulating Gene Expression, Embryonic Development, Cell Differentiation, Maintaining Genome Stability, and Other Biological Processes, as Well as in Disease Progression. Precise and Comprehensive Analysis of Whole-Genome DNA Methylation Patterns Is of Inestimable Importance for Deeply Understanding the Biological Functions of DNA Methylation, and for Discovering Potential Biomarkers for Disease Diagnosis and Therapeutic Targets.
From early specific site detection to today's whole-genome scale analysis, methylation detection technology has undergone revolutionary development. A variety of whole-genome DNA methylation detection technologies have emerged, such as Whole-Genome Bisulfite Sequencing (WGBS), Illumina Methylation Array (EPIC, MSA), Enzymatic Methyl-seq (EM-seq), and Oxford Nanopore Technologies' third-generation sequencing (ONT). This document summarizes the latest comparative studies from 2023–2025 on mainstream DNA methylation detection technologies such as Illumina Methylation Array, WGBS, EM-seq, and ONT, focusing on real experimental results and firsthand comparative literature evidence to provide reliable academic support for suppliers and partners.
Based on a multicenter systematic comparative study published in *Epigenetics & Chromatin* in 2025, parallel experimental analyses of human tissues, cell lines, and blood samples revealed that EM-seq and WGBS exhibit the highest consistency (Fleiss' kappa=0.78, correlation r>0.93). Moreover, EM-seq demonstrates more uniform coverage in GC-rich regions, significantly reducing the GC bias inherent to WGBS. The Illumina EPIC array achieves moderate-to-high agreement (kappa≈0.65–0.78) with WGBS/EM-seq at overlapping CpG sites (Figure 1), making it suitable for large-scale cohort studies [1]. In contrast, although ONT shows slightly lower overall agreement with WGBS, the R10 chip significantly improves accuracy (correlation with WGBS r=0.969), particularly providing more reliable detection in extreme methylation regions (0%-10% and 90%-100%), thus being more appropriate for exploratory research [2]. Therefore, despite differences in chemical principles and resolution among these methods, cross-platform consistency in capturing methylation dynamics is evident. This provides a technical basis for research: sequencing methods (such as WGBS, EM-seq, and ONT) are better suited for discovering novel methylation sites and mechanisms, while Illumina methylation array technology, with its high reproducibility, low cost, and standardized advantages, is currently the most widely applied in large-scale cohort validation and translational studies.
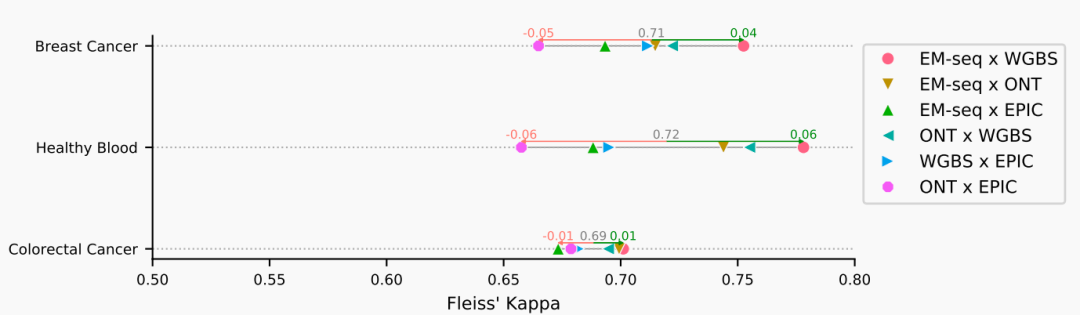
Figure 1 Consistency Analysis Among Technologies
Under the condition of coverage ≥10×, each technology detected unique sites—ONT exclusively covered 5.1 million CpG sites (83% located in complex regions such as intergenic areas), EM-seq and WGBS respectively identified 850,000 and 622,000 unique sites, while the EPIC chip also detected 34,700 sites not covered by the other three platforms.[1]. This "unique coverage" is not a flaw but rather highlights the advantages of each method. In practical applications, Riccardo E. Marioni's team at the University of Edinburgh, UK, used the Illumina EPIC array in a large cohort of 17,865 individuals, explaining 50.0% of the epigenetic variance associated with smoking pack-years. They identified 42 independently associated CpG sites and developed the biomarker mCigarette (AUC=0.98), enabling large-scale screening and biomarker development.[3]Based on this, TWIST targeted sequencing and ONT long-read sequencing further identified several novel loci (such as CNTNAP2) in small samples and compensated for the detection blind spots in high GC regions. However, these new findings still require statistical validation in large cohorts using arrays. This has led to a complementary model of "microarrays responsible for broad screening and validation—sequencing methods aiding in-depth discovery," providing a solid technical pathway for comprehensively elucidating smoking-related methylation mechanisms.
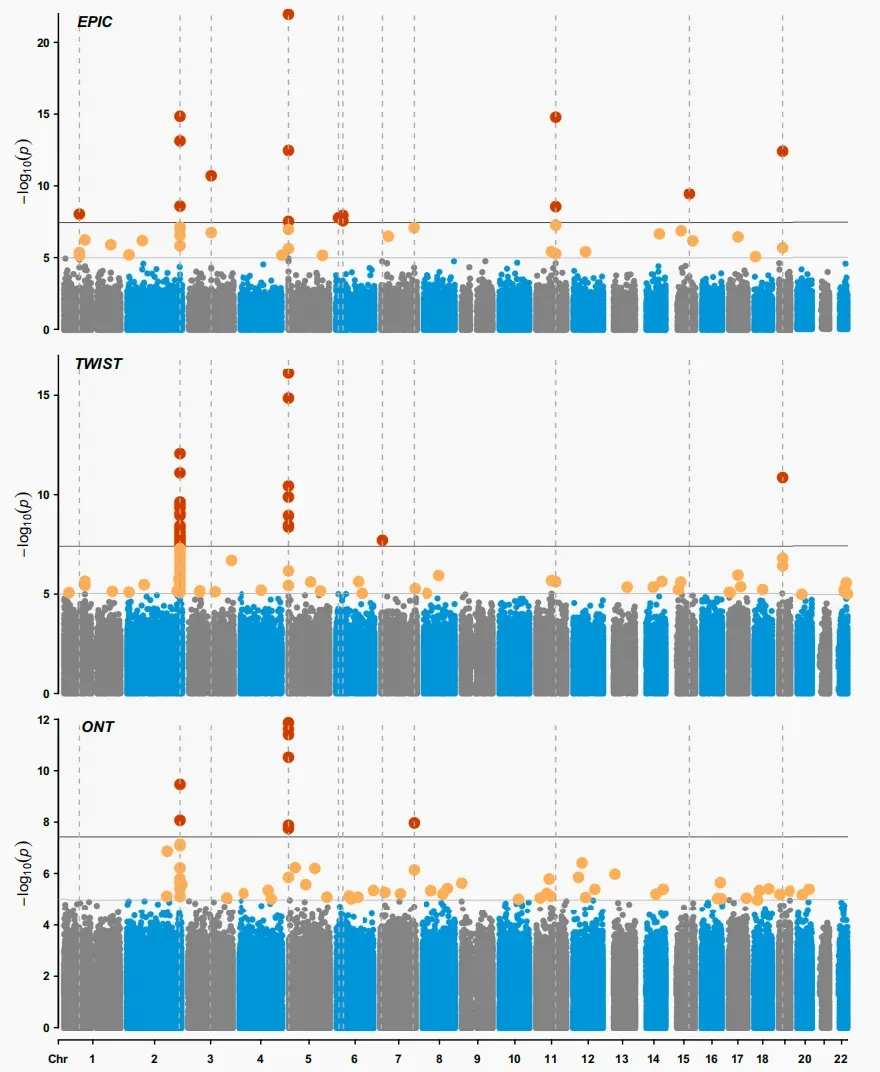
Figure 2: Genome-wide epigenetic association study of smokers and non-smokers in the GS cohort
Under the condition of coverage ≥10×, ONT can detect approximately 51 million CpG sites, EM-seq about 46.7 million, and WGBS about 44.4 million; whereas EPIC has approximately 866,000 effective probes. WGBS has the highest number of unique missing sites, suggesting its inadequacy in spacer and repetitive sequence regions. Specifically, WGBS fails to detect these sites due to the degradation of unmethylated C-rich sequences (e.g., CCCTAA telomeric repeats) caused by bisulfite treatment. In contrast, EM-seq and ONT miss fewer sites.[1]。
The "unique coverage" of different methods mainly reflects the differences brought by their chemical principles or read length characteristics. Among the uniquely covered sites, ONT detects the most (about 4.17 million), followed by EM-seq (833,000) and WGBS (594,000). These unique sites are mostly located in intergenic regions and repetitive regions, where long-read methods can span such areas, while short-read methods are more prone to missed detections. Overall, different platforms complement each other in detection scope: sequencing methods excel in breadth and coverage of complex regions, while predefined probe platforms focus on functional regions, making them more conducive to data integration across studies.
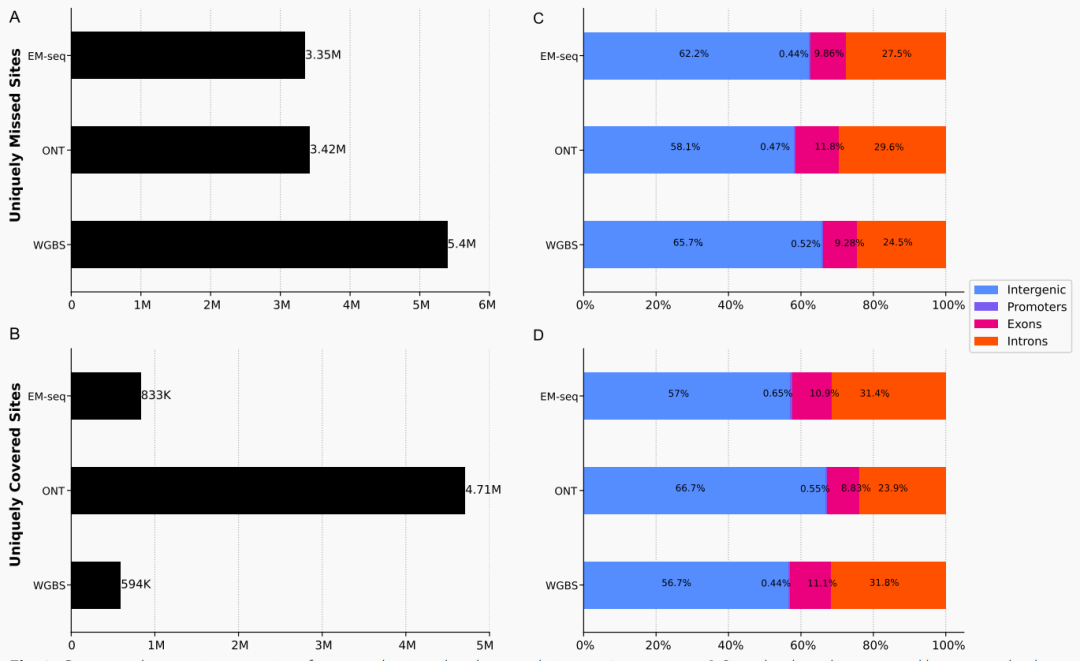
Figure 3 Count and genomic annotation of unique omissions and covered sites at 10X coverage
Traditional WGBS converts unmethylated C to U through bisulfite treatment, with a conversion efficiency typically exceeding 99%. However, bisulfite treatment may cause DNA degradation, affecting the actual conversion efficiency. EM-seq uses an enzymatic reaction to convert unmethylated C to U, with a conversion efficiency close to that of WGBS (e.g., EM-seq’s conversion efficiency for unmethylated λ is 99.46%). Moreover, EM-seq provides better coverage in GC-rich regions [1], avoiding the GC bias present in WGBS, making it more suitable for degraded samples such as FFPE/cfDNA.
A 2024 study published in *BMC Genomics* systematically evaluated the performance of four mainstream DNA methylation detection technologies in GC-rich regions using the same batch of human blood samples (Figure 4). The results showed that WGBS exhibited a sharp decline in coverage and systematically overestimated methylation levels in extremely high GC regions (>75%), requiring μg-level input. In contrast, EM-seq, which uses optimized enzymatic conversion (TET2/APOBEC), significantly reduced DNA damage, provided more uniform coverage and accurate reads in GC-rich regions, with a minimum input as low as 10–25 ng, delivering superior data quality but requiring longer library preparation time. ONT detects methylation directly without conversion, and its coverage is completely unaffected by GC content (due to long-read characteristics); in GC-rich regions, its methylation readings are closer to those of EM-seq. The Illumina EPIC array, following bisulfite treatment, achieves a 99% conversion rate and technical reproducibility (r > 0.98), but shows slightly higher readings than sequencing methods in extremely high GC regions due to probe cross-reactivity [4]. Overall, EM-seq excels in low-input scenarios and GC bias control, ONT provides unbiased coverage uniformity, while Illumina EPIC and WGBS offer practical advantages in cost-effectiveness and large-scale data comparison, respectively.
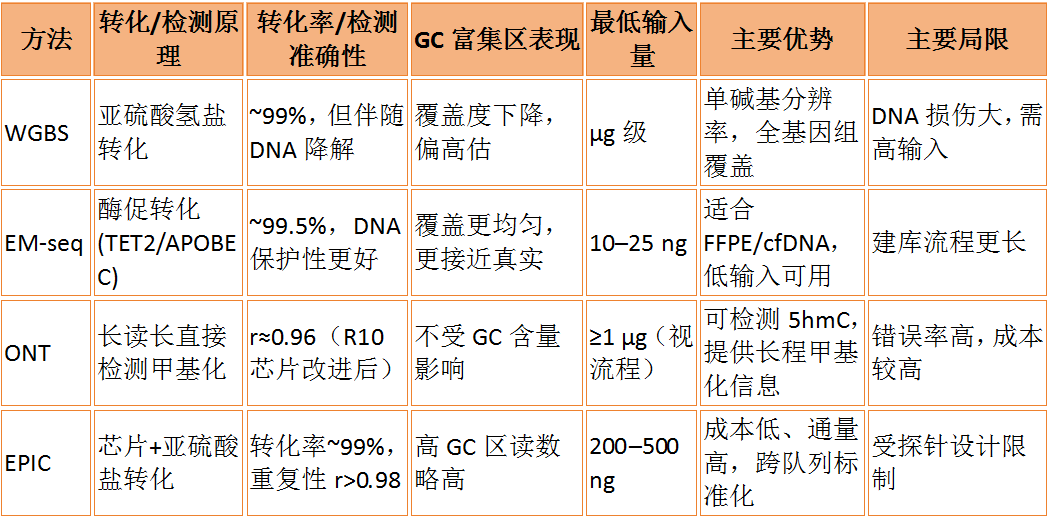
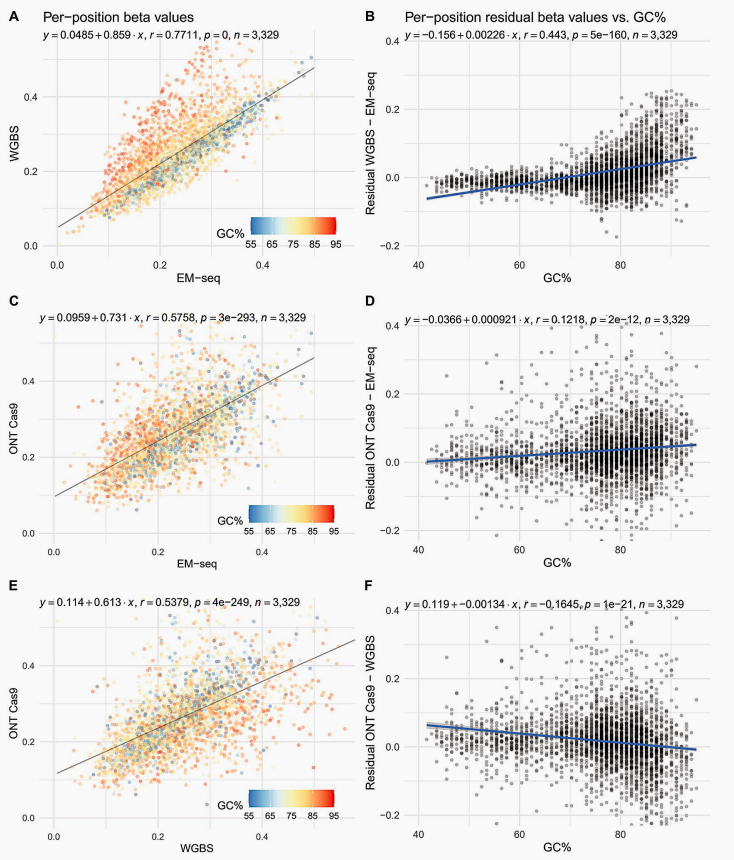
Figure 4 Pairwise comparison of methylation levels at each site on the 45S locus
Notably, the latest research shows that the EPIC v2 chip also exhibits excellent compatibility with special sample types. Karolinska Institutet randomly selected seven dried blood spot (DBS) samples from newborns born between 1985 and 2003 to evaluate the performance of the Illumina EPIC v2 chip in such samples. The results showed that, despite the long storage time of the samples, the probe call rate reached 99.42%–99.89% at a threshold of p<0.01 and 99.72%–99.93% at p<0.05 (Figure 5). This indicates that the Illumina EPIC v2 can still achieve high-quality methylation data under conditions of low DNA input (19.2–99.2 ng) and long-term storage, providing new possibilities for large-scale longitudinal studies.[5]。
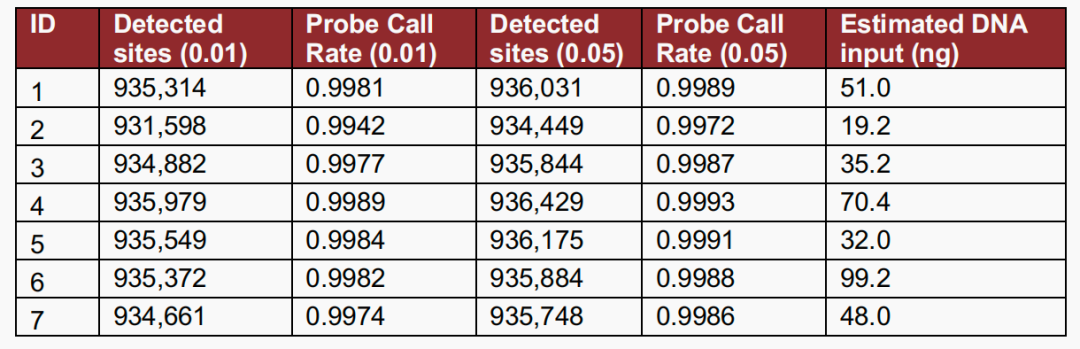
Figure 5 Number of DNA methylation sites at different detection levels for each pilot sample
Traditional WGBS cannot distinguish between 5mC and 5hmC, but when combined with oxidative bisulfite sequencing, it enables the detection of 5hmC. Its weak long-read capability makes it difficult to span complex regions when detecting continuous large methylation patterns, and it is susceptible to GC content, leading to uneven coverage. ONT can directly detect 5hmC and provides continuous methylation information (long-range information) in repetitive sequences, telomeres, and large segments due to its long-read capability, but it has a relatively high error rate in 5hmC quantification. EM-seq can also differentiate between 5mC and 5hmC and is suitable for handling minimal sample quantities. The Illumina MSA chip offers unique advantages in large-scale studies: its design covers a large number of functional sites related to 5hmC, supporting epigenetic clock and trait association studies in large cohorts. A recent report in *Cell Genomics* describes the use of the MSA chip combined with an innovative bisulfite-APOBEC3A conversion strategy to achieve ternary resolution of 5mC, 5hmC, and unmodified cytosine, constructing high-throughput 5hmC maps for 117 tissue types, revealing its enrichment characteristics in the nervous system and negative correlation with proliferation rates [6]. Although chips cannot provide continuous fragment information at the single-molecule level like long-read sequencing, the MSA chip has become one of the few methods capable of systematically examining 5hmC in large-scale studies by incorporating numerous functional sites associated with 5hmC into its design.
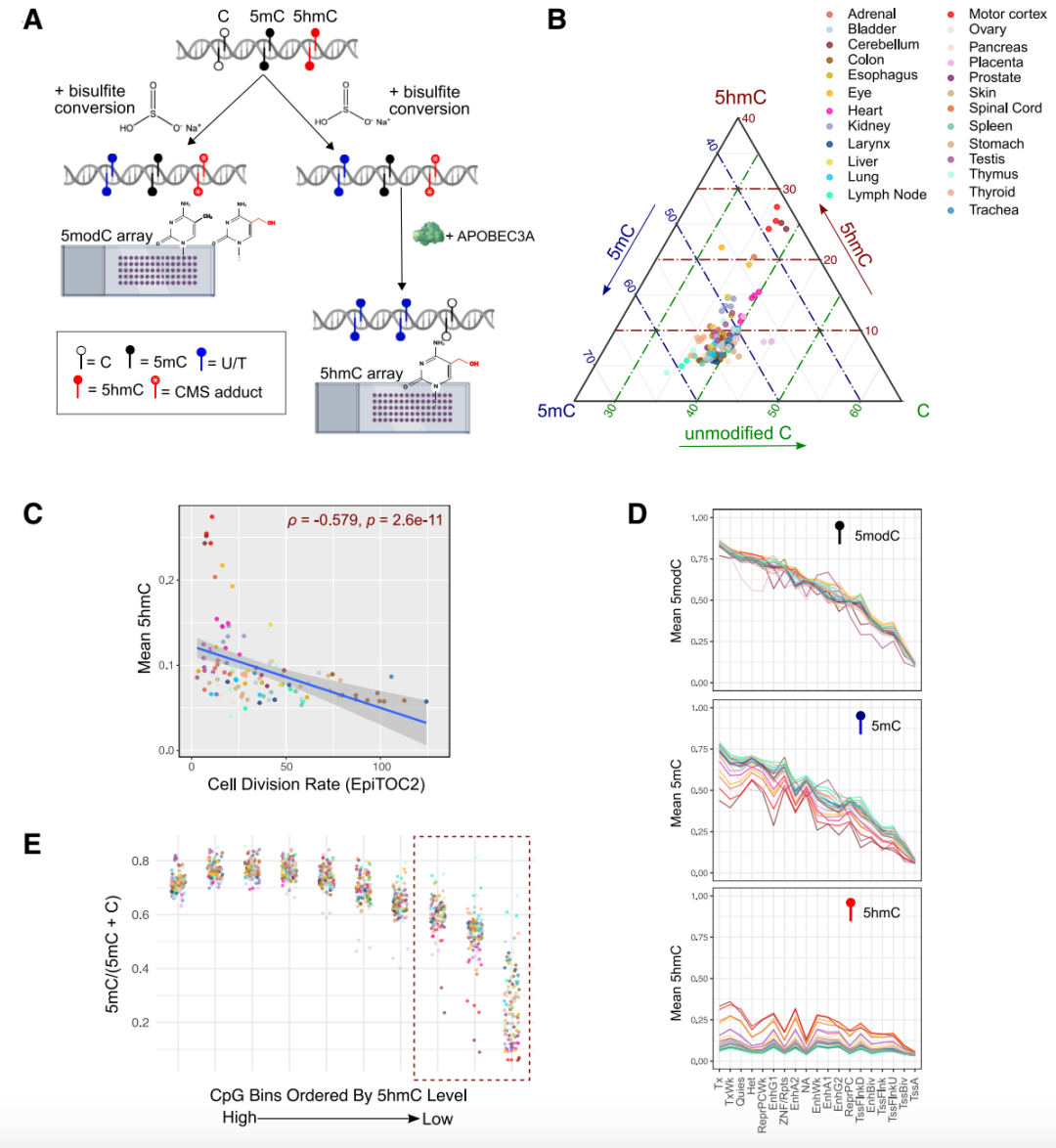
Figure 6 Global 5hmC in human tissues analyzed by Illumina-MSA
WGBS, as the gold standard technology for DNA methylation analysis, provides a whole-genome methylation map at single-base resolution, enabling precise detection across the entire genome. It holds irreplaceable value in epigenetic research, suitable for discovering new methylation biomarkers, studying genome-wide methylation dynamics during development and disease processes, and constructing reference methylation maps. In cancer research, WGBS can comprehensively reveal tumor-specific methylation patterns, providing crucial evidence for understanding tumorigenesis mechanisms and developing diagnostic markers.
EM-seq as a Key Tool for Methylation Detection in Trace Samples: Reducing DNA Input to an Ultra-Low Level of 10ng for Comprehensive Methylation Analysis of Precious Samples. A study published in *Briefings in Bioinformatics* focuses on high-risk neuroblastoma, combining liquid biopsy with EM-seq to construct DNA methylation and genomic profiles using trace plasma-derived DNA. The characteristic features of cfDNA methylation are used to predict genomic mutation subtypes in neuroblastoma patients and further analyze patient prognosis.[7]。
Illumina Methylation Array, as the core tool for preset probe-targeted methylation research, features a high-throughput and targeted design based on hybridization probes. It can simultaneously detect hundreds of thousands of pre-selected specific CpG sites at an extremely low cost per sample. In most practical research applications, this technology remains the preferred solution in the field. Illumina-EPIC v2 expands to 930,000–940,000 CpGs, covering over 99% of RefSeq genes, targeting cancer driver genes, CpG islands, promoters, enhancers, and transcription regulatory regions, making it more suitable for multi-ancestry populations and enhancer region studies.[8]A study published in Epigenetics Communications showed that Illumina-EPIC v2 maintains a methylation quantification accuracy of ρ > 0.91 with 1ng DNA input. It also includes 183k new enhancer probes and 824 cancer mutation NV probes, enabling a "methylation-mutation-copy number" triple assay. This has been validated in multi-ancestry populations and DNMT knockout models, demonstrating high coverage and reproducibility in large cohort and clinical translational studies [9]. Illumina-MSA focuses on "functional site targeting + high-throughput screening," optimized for million-person cohorts, balancing throughput and cost. It covers approximately 270,000 selected CpG sites related to human traits, diseases, aging, and environmental exposure, supporting 5mC/5hmC/C ternary detection. Compatible with sample types such as blood and frozen DNA, it offers high reproducibility, low cost, and high sample throughput (48 samples per chip), but does not support FFPE samples. It is suitable for research tasks like methylation risk scoring (MRS), tissue localization, immune profiling, and epigenetic clock construction.[6]。
ONT, as a cutting-edge tool for real-time detection of DNA methylation at the single-molecule level, can span complex regions and retain the original methylation signals due to its long-read direct sequencing. It shows potential in analyzing continuous large-scale methylation patterns and haplotypes. According to a report in *Nature Medicine*, a team from Heidelberg University in Germany developed the Rapid-CNS2 platform based on ONT, which can provide methylation classification and copy number information within approximately 30 minutes during surgery and complete comprehensive molecular subtyping within 24 hours, with 94.6% consistency compared to traditional methods.[9]. These results demonstrate the exploratory potential of ONT in real-time applications. However, it is important to emphasize that in international practice for central nervous system tumor classification, the universally validated standard method, long advocated by Heidelberg, remains the molecular classification process based on the Illumina methylation array. Its accuracy and reproducibility have been widely recognized, making it an essential reference in the current WHO CNS classification.[10]。
In light of the current research landscape, strategies for selecting DNA methylation detection methods should be based on research objectives and sample characteristics: sequencing methods (e.g., WGBS, EM-seq, ONT) excel in discovering new sites and analyzing complex regions, while targeted capture methods like TWIST can provide focused supplementation in functional regions. However, when advancing to large-scale validation and translational stages, reliance on the advantages of Illumina chips (e.g., EPIC v2, MSA) in high-throughput, low-cost, and standardized applications remains essential to achieve feasibility for larger-scale studies.
01 Large-Scale Studies: For EWAS or longitudinal cohort studies involving tens of thousands of participants, it is recommended to use Illumina's EPIC v2 or MSA chips. These two options, with their high reproducibility and low cost, can efficiently support large-scale methylation screening and phenotype association studies.
02 Complex Regions and Modification Differentiation: If the research focus is on complex regions of the genome (such as repetitive sequences, high GC regions) or requires precise differentiation between 5mC and 5hmC, methods like EM-seq or ONT sequencing can be employed for exploration. In terms of large-scale applications for 5hmC, the Illumina MSA chip, with its pre-designed functional site layout, has become the only tool capable of systematically incorporating 5hmC in large cohorts.
03 Low Input and Degraded Samples: EM-seq maintains good performance with a starting amount of 10–25ng, making it the preferred choice for cfDNA and FFPE samples; while the EPIC v2 chip, despite requiring a higher standard input, achieved a probe call rate of >99.4% in real-world samples such as neonatal dried blood spots, demonstrating certain tolerance and practical value.
04 Overall Strategy: From the perspective of research efficiency and transformation value, it is recommended to adopt a two-phase approach of "discovery sequencing + large-scale chip screening": first, use technologies such as WGBS, EM-seq, or ONT to explore potential methylation-associated sites and functional regions in small-scale studies, then validate and promote them in large populations through EPIC v2 or MSA chips, achieving efficient integration from scientific discovery to clinical and public health applications.
References: (Swipe to view)
[1]de Abreu AR, Ibrahim J, Lemonidis V, et al. Comparison of current methods for genome-wide DNA methylation profiling. Epigenetics Chromatin. 2025 Aug 25;18(1):57. doi: 10.1186/s13072-025-00616-3.
[2]Genner R, Akeson S, Meredith M, et al. Assessing DNA methylation detection for primary human tissue using Nanopore sequencing. Genome Res. 2025 Apr 14;35(4):632-643. doi: 10.1101/gr.279159.124.
[3]Chybowska AD, Bernabeu E, Yousefi P, et al. A blood- and brain-based EWAS of smoking. Nat Commun. 2025 Apr 4;16(1):3210. doi: 10.1038/s41467-025-58357-6.
[4]Guanzon D, Ross JP, Ma C, et al. Comparing methylation levels assayed in GC-rich regions with current and emerging methods. BMC Genomics. 2024 Jul 30;25(1):741. doi: 10.1186/s12864-024-10605-7.
[5]Brander, Gustaf & Karlsson, Håkan & Dalman, et al. (2025). Performance of the Illumina Infinium MethylationEPIC v.2 array with low DNA input from Swedish neonatal dry blood spots. 10.1101/2025.03.10.25323486.
[6]Goldberg DC, Cloud C, Lee SM, et al. Scalable screening of ternary-code DNA methylation dynamics associated with human traits. Cell Genom. 2025 Jul 2:100929. doi: 10.1016/j.xgen.2025.100929. Deacon S, Cahyani I, Holmes N, et al. ROBIN: A unified nanopore-based assay integrating intraoperative methylome classification and next-day comprehensive profiling for ultra-rapid tumor diagnosis. Neuro Oncol. 2025 May 20:noaf103. doi: 10.1093/neuonc/noaf103.
[7]Noguera-Castells A, García-Prieto CA, Álvarez-Errico D, et al. Validation of the new EPIC DNA methylation microarray (900K EPIC v2) for high-throughput profiling of the human DNA methylome. Epigenetics. 2023 Dec;18(1):2185742. doi: 10.1080/15592294.2023.2185742.
[8]Kaur D, Lee SM, Goldberg D, et al. Comprehensive Evaluation of The Infinium Human MethylationEPIC v2 BeadChip. Epigenetics Commun. 2023;3(1):6. doi: 10.1186/s43682-023-00021-5.
[9]Patel A, Göbel K, Ille S, et al. Prospective, multicenter validation of a platform for rapid molecular profiling of central nervous system tumors. Nat Med. 2025 May;31(5):1567-1577. doi: 10.1038/s41591-025-03562-5.
[10]Capper D, Jones DTW, Sill M, et al. DNA methylation-based classification of central nervous system tumours. Nature. 2018;555(7697):469-474. doi:10.1038/nature26000
This article is a reprint, source: Sequencing China.If there is any infringement or reprint restriction on the subscription account, please contact us (or leave a message below the official account), and we will contact you promptly and proceed with deletion.
