TIDES: Beyond Weight Loss and GLP-1, the Peptide World Is Full of Exciting Developments

MannKind
Developer of Diabetes Treatment Products

Sciwind
Metabolic Disease Drug Developer

Novo Nordisk
Insulin Developer and Manufacturer

Warm Tips
Peptide Research Society Builds Reader Communication Group~
Industry Exchange, Business Cooperation, Report Consultation
Please add the editor's WeChat for subsequent group joining.

Research Progress
MannKind
California, USA, October 13, 2025 – MannKind (NASDAQ: MNKD) announced that the FDA has accepted its supplemental Biologics License Application (sBLA) for the approval of Afrezza (inhaled human insulin powder) for use in children and adolescents aged 4 to 17 with type 1 or type 2 diabetes. The PDUFA goal review date for this application is May 29, 2026.
If approved, Afrezza will become the first needle-free insulin treatment option for children in over 100 years, providing an alternative to multiple daily injections or insulin pumps. The application is based on data from the Phase 3 INHALE-1 study. This 26-week randomized open-label clinical trial evaluated the efficacy and safety of Afrezza combined with basal insulin compared to multiple daily injections (MDI) combined with basal insulin and included 26 weeks of extended safety data, during which all remaining MDI patients switched to Afrezza. Top-line six-month data from INHALE-1 was released in December 2024, with full results to be presented at the International Society for Pediatric and Adolescent Diabetes (ISPAD) conference in November.
Afrezza has been approved by the FDA for use in adults (over 18 years old) since June 2014, and it has also been approved in India and Brazil, being incorporated into the American Diabetes Association (ADA) Standards of Care.
Sciwind
On 2025-10-13, Sciwind's Enoxaglutide Tablets received clinical trial approval (CXHL2500742) from the CDE for weight management in individuals with obesity or overweight who have at least one weight-related comorbidity.
Federal Biosciences
On 2025-10-13, UBT251 Injection from Federal Biotech registered four clinical trials in the CTR. This is a pharmacokinetic study evaluating UBT251 Injection in subjects with normal renal function and subjects with renal impairment.
UBT251 is a tri-target receptor agonist independently developed by Federal Pharmaceutical, targeting the three hormone receptors GLP-1, GIP, and GCGR. In June this year, Novo Nordisk paid USD 180 million (approximately RMB 1.293 billion) upfront to Zhuhai Federal Pharmaceutical in China for the global rights outside Greater China to its self-developed triple-target weight loss drug UBT251.
Shiyao Group Baike
On 2025-10-14, the marketing application for Edaglutide α Injection, a Class 1 biologic drug developed by Baike of CSPC Group, was accepted by the CDE (CXSS2500109, CXSS2500110, CXSS2500111).
Chengdu Tongde Pharmaceutical Co., Ltd.
On 2025-10-14, the clinical application of sodium colistimethate injection from Chengdu Tongde Pharmaceutical was accepted by CDE (CYHL2500180).
Hunan Kelun
On 2025-10-14, Hunan Kelun's cyclosporine oral solution marketing application was accepted by the CDE (CYHS2503654).
Fujian Shengdi Pharmaceutical
On 2025-10-15, the clinical application for HRS9531 injection, a GLP-1/GIP dual receptor agonist developed by Fujian Shengdi Pharmaceutical (parent company: Hengrui Medicine), was accepted by the CDE.
Zhongshan MannKind Pharmaceutical
On 2025-10-15, the clinical application of Semaglutide Injection by Zhongshan Wanhhan Pharmaceutical was accepted by CDE.
Shenzhen Carbon Cloud Intelligent Peptide
On 2025-10-15, the clinical application of Shenzhen Carbon Cloud Intelligent Peptide's antimicrobial peptide iCAMP016 vaginal gel was accepted by the CDE.
Lilly
U.S. 2025-10-15, Lilly announced that its oral small molecule GLP-1 receptor agonist Orforglipron met the primary endpoints in both Phase III ACHIEVE-2 and ACHIEVE-5 studies, significantly reducing glycated hemoglobin (A1C) levels in patients with type 2 diabetes.
In the ACHIEVE-2 study, after 40 weeks of treatment with Orforglipron (3 mg, 12 mg, 36 mg), A1C decreased by up to 1.7%, significantly outperforming the control drug dapagliflozin (-0.8%), successfully achieving the primary endpoint. In the ACHIEVE-5 study, Orforglipron combined with insulin glargine also significantly improved blood glucose control, with a maximum A1C reduction of 2.1%, showing a statistically significant difference compared to placebo (-0.8%) (p<0.001).
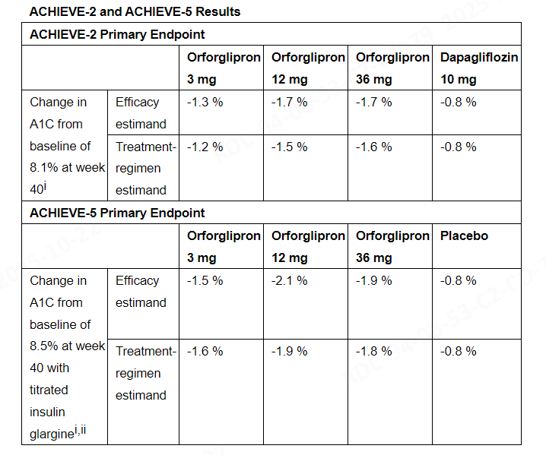
In two studies, Orforglipron achieved significant weight loss and improvements in multiple cardiovascular risk factors, consistent with previous research findings. In terms of safety, the most common adverse reactions were mild to moderate gastrointestinal effects, which were well-tolerated with no signals of hepatotoxicity.
ProteinQure
Toronto, Canada, October 21, 2025 – Biotech company ProteinQure announced that its self-developed peptide-oligonucleotide conjugation technology successfully achieved efficient delivery of small interfering RNA (siRNA) to the central nervous system (CNS) in non-human primates. The study utilized intrathecal administration and achieved widespread distribution of siRNA in the brain through receptor-mediated transport mechanisms.
In the 28-day study, ProteinQure's conjugated molecules were directly compared to the current clinically leading lipid-based delivery systems, showing best-in-class delivery efficiency and demonstrating superior delivery advantages in specific cell types.
The company will present more early data on its blood-brain barrier crossing technology project at the 21st Annual Meeting of the Oligonucleotide Therapeutics Society (OTS), to be held in Budapest in October 2025.
Pulai Pharmaceuticals
On October 16, 2025, Jiangsu Pulaite Pharmaceuticals announced the completion of the first participant dosing in the Phase I clinical trial of its Category 1 new drug, antimicrobial peptide PL-3301 oral gel. Safety follow-up for study participants is currently ongoing. This study involves healthy adult participants and is conducting a single-dose escalation clinical trial to evaluate the safety, tolerability, and pharmacokinetic (PK) characteristics of the antimicrobial peptide PL-3301 oral gel, providing a basis for subsequent clinical development. The study has received approval from the NMPA and is actively progressing with various clinical research activities.
PL-3301 Antimicrobial Peptide Oral Gel is a novel peptide-based broad-spectrum anti-infective drug, classified as a first-in-class non-antibiotic anti-infective. It features a unique antifungal and antibacterial mechanism of action. Preclinical study data supports its progression into clinical development, aiming to provide a new treatment method for oropharyngeal candidiasis.
PeptiDream
Kawasaki, Japan, 2025-10 – PeptiDream Inc. (Tokyo Stock Exchange: 4587) announced that its wholly-owned subsidiary PDRadiopharma has jointly launched a pivotal Phase II clinical trial (jRCT: 2031250225) in Japan with Curium Group, a leader in nuclear medicine, for the PET imaging diagnosis of prostate cancer patients using 64Cu-PSMA-I&T.
64Cu-PSMA-I&T is a copper-64 labeled positron emission tomography (PET) radiopharmaceutical that targets prostate cancer cells expressing prostate-specific membrane antigen (PSMA). The development of this drug is being advanced in parallel with its therapeutic counterpart, 177Lu-PSMA-I&T, together forming a "theranostic radiopharmaceutical companion pair."
This open-label, single-arm Phase II study will evaluate the sensitivity, specificity, and safety of 64Cu-PSMA-I&T, with plans to recruit approximately 70 patients newly diagnosed with intermediate-, high-, or very-high-risk prostate cancer who are scheduled to undergo radical prostatectomy and pelvic lymph node dissection. The study will be conducted in Japan as a registrational trial and will utilize bridging data from Curium’s global clinical program. Meanwhile, clinical research on 177Lu-PSMA-I&T for the treatment of castration-resistant metastatic prostate cancer (mCRPC) is also being prepared.
Currently, Curium's 177Lu-PSMA-I&T has achieved the primary endpoint in the global Phase III ECLIPSE study (NCT05204927), demonstrating significant clinical benefits; meanwhile, 64Cu-PSMA-I&T has also validated its diagnostic accuracy in multiple international studies (SOLAR series), laying the foundation for its registration in Japan.
Yunnan Sciwind Pharmaceutical
On 2025-10-17, the clinical application of Teriparatide Injection from Yunnan Xianshi Pharmaceutical was accepted by CDE (CXHL2501135).
Novo Nordisk
[US & Denmark, 2025-10-17] Novo Nordisk announced that the FDA has approved its oral GLP-1 receptor agonist Rybelsus® (7 mg and 14 mg) for reducing the risk of major adverse cardiovascular events (MACE) in adults with type 2 diabetes at high risk, including cardiovascular death, myocardial infarction, and stroke. Rybelsus® has thus become the only FDA-approved oral GLP-1 drug with a cardiovascular risk reduction effect, suitable for both primary and secondary prevention of cardiovascular events.
In the Phase IIIb SOUL study, oral semaglutide 14 mg significantly reduced the risk of MACE by 14% (HR=0.86, p=0.006) when added to standard care. Compared with placebo, the incidence rates of cardiovascular events in participants over four years were 12.0% and 13.8%, respectively. The study also showed that the treatment had a good safety profile, with common adverse reactions being mild to moderate gastrointestinal effects.
Currently, Novo Nordisk has submitted a supplemental application in the United States for the once-daily oral version of Wegovy® (semaglutide) for the treatment of obesity, with an approval decision expected within the year.
ITM
Berlin, Germany, October 18, 2025 – ITM Isotope Technologies Munich SE (ITM), a radiopharmaceutical biotech company, presented the latest results of the Phase III COMPETE (NCT03049189) study at the 2025 European Society for Medical Oncology (ESMO) Annual Congress: In patients with Grade 1 or 2 somatostatin receptor (SSTR)-positive gastroenteropancreatic neuroendocrine tumors (GEP-NETs), non-carrier 177Lu-edotreotide (ITM-11) demonstrated a significantly higher objective response rate (ORR) and longer median progression-free survival (mPFS) compared to everolimus. The efficacy advantage was observed across multiple patient subgroups. This global multicenter Phase III study compares the efficacy and safety of 177Lu-edotreotide versus everolimus in patients with unresectable, progressive G1/G2 GEP-NETs and has met its primary endpoint, confirming that 177Lu-edotreotide significantly improves progression-free survival.
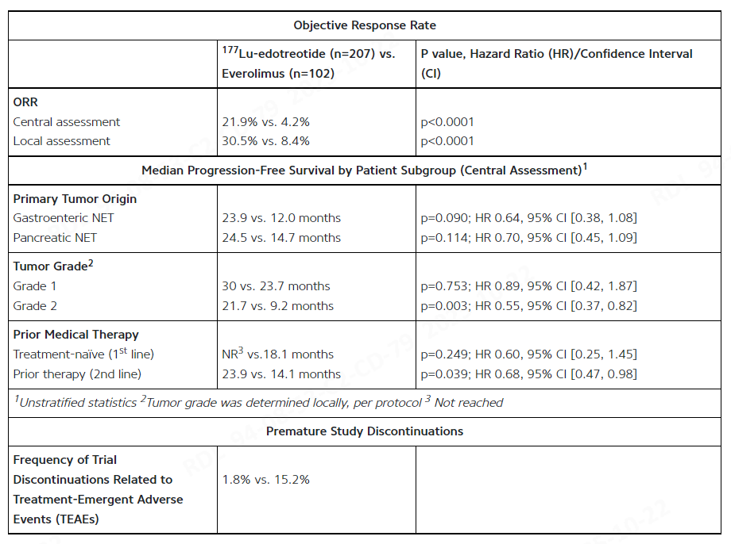
The COMPETE study enrolled 309 patients, who were randomly assigned in a 2:1 ratio to the 177Lu-edotreotide group (n=207) or the everolimus group (n=102). The study was evaluated by an independent blinded central review committee (BICR). The results showed that the mPFS in the 177Lu-edotreotide group was significantly better than that in the everolimus group (p<0.022). Subgroup analysis revealed:
- ORR (Central Assessment) was 21.9% vs. 4.2%; Local Assessment was 30.5% vs. 8.4% (both p<0.0001);
- mPFS in gastrointestinal NET and pancreatic NET reached 23.9 vs. 12.0 months and 24.5 vs. 14.7 months, respectively;
- mPFS was significantly prolonged in Grade 2 tumor patients (21.7 vs. 9.2 months, p=0.003);
- The rate of premature discontinuation due to treatment-related adverse events was only 1.8%, significantly lower than the 15.2% in the everolimus group.
The COMPETE dosimetry data presented at the EANM (European Association of Nuclear Medicine) annual congress showed that 177Lu-edotreotide enables precise radiation delivery, significantly reducing radiation exposure to healthy tissues. The report received the Marie Curie Award in recognition of its outstanding scientific contribution to the field of nuclear medicine.
Pfizer & Astellas
United States, 2025-10-18 — Pfizer and Astellas today announced the final results of the Phase III EMBARK study: In men with non-metastatic hormone-sensitive prostate cancer (nmHSPC) and high-risk biochemical recurrence (BCR), XTANDI® (enzalutamide) in combination with leuprolide significantly reduced the risk of death and extended overall survival (OS). The results were presented as an oral report at the 2025 European Society for Medical Oncology (ESMO) Annual Meeting and simultaneously published in The New England Journal of Medicine.
The study showed that the 8-year survival rate was 78.9% in the XTANDI® plus leuprolide group, compared to 69.5% in the leuprolide-alone group, with combination therapy reducing the risk of death by 40.3% (HR=0.597, 95% CI: 0.444–0.804, p=0.0006). Although XTANDI monotherapy demonstrated a trend toward improved survival, it did not reach statistical significance (HR=0.83, p=0.1867). This makes XTANDI® the first and only androgen receptor inhibitor regimen proven to provide an overall survival benefit in this population.
In the EMBARK study, with a median follow-up of approximately 94 months, the safety profile of the XTANDI combination group was consistent with previous analyses, and no new safety signals were identified. The most common adverse reactions included hot flashes and fatigue. The primary analysis published earlier (in 2023) demonstrated that the combination of XTANDI and leuprolide significantly extended metastasis-free survival (MFS), reducing the risk by 58% compared to leuprolide alone (HR=0.42, p<0.001).
Novartis
Switzerland, 2025-10-19 — Novartis presented the results of the Phase III PSMAddition study at the Presidential Symposium of the 2025 European Society for Medical Oncology (ESMO) Congress: In patients with PSMA-positive metastatic hormone-sensitive prostate cancer (mHSPC), Pluvicto™ (177Lu vipivotide tetraxetan) combined with standard treatment (ARPI + ADT) significantly reduced the risk of radiographic progression or death by 28% (HR=0.72, 95% CI: 0.58–0.90) compared to standard treatment alone, and showed a trend toward overall survival (OS) improvement (HR=0.84).
The study also found that the complete remission rate in the Pluvicto group was 57.1% (42.3% in the control group), and the overall response rate (ORR) was 85.3% (80.8% in the control group), significantly delaying disease progression to metastatic castration-resistant prostate cancer (mCRPC) (HR=0.70). The efficacy advantage was consistent across all predefined subgroups.
In terms of safety, the spectrum of adverse events in the Pluvicto group was consistent with previous PSMAfore and VISION studies, with no new safety signals identified. The incidence of grade 3 or higher adverse events was 50.7% (43% in the control group), with the most common being dry mouth, fatigue, nausea, hot flashes, and anemia.
PSMAddition is the third Phase III study of Pluvicto to yield positive results. Following the FDA approval of Pluvicto in March 2025 for mCRPC prior to chemotherapy, this result further demonstrates its potential clinical value in earlier-stage metastatic prostate cancer. Novartis plans to submit a regulatory application by the end of the year, and if approved, it would double the number of potentially eligible patients for Pluvicto.
Ascletis Pharma
Hong Kong, 2025-10-19 – Ascletis Pharma (Ascletis, HKEX: 1672) announced that the Phase IIa clinical trial in the United States for its once-monthly subcutaneous injection sustained-release formulation of the small molecule GLP-1 receptor agonist ASC30 (ASC30 SQ depot) has completed full enrollment, with a total of 65 subjects, all of whom are obese or overweight individuals with weight-related comorbidities.
This 12-week randomized, double-blind, placebo-controlled multicenter study aims to evaluate the safety, tolerability, and weight-loss efficacy of ASC30 SQ depot. The study includes three dose groups, with top-line data expected to be released in Q1 2026.
Previously, in the Phase Ib study, ASC30's ultra-long-acting sustained-release formulation demonstrated an observed half-life of 46 days (with a terminal half-life of 36 days) in obese subjects, supporting a once-monthly dosing regimen. Its peak-to-trough concentration ratio was approximately 1.5:1, indicating stable pharmacokinetic characteristics. This formulation is based on Ascletis' proprietary ultra-long-acting sustained-release platform (ULAP), which does not rely on albumin to extend the half-life, thus overcoming the traditional bottleneck where peptide drugs are limited to a half-life of approximately 20 days.
Apellis Pharma
Massachusetts, USA, October 20, 2025 – Apellis Pharmaceuticals (NASDAQ: APLS) announced new data from the open-label phase of the Phase III VALIANT study of its C3 inhibitor EMPAVELI® (pegcetacoplan), demonstrating significant and sustained efficacy in patients with C3 glomerulopathy (C3G) and primary immune complex membranoproliferative glomerulonephritis (IC-MPGN).
The study results showed that EMPAVELI maintained a significant reduction in proteinuria regardless of whether patients received concomitant immunosuppressive therapy or their baseline proteinuria levels, with an approximately 68% decrease in proteinuria from baseline at one year (p<0.0001). About one-third of patients achieved complete proteinuria remission (UPCR≤0.5 g/g), and the effect persisted for one year. The glomerular filtration rate (eGFR) remained stable, and the safety profile was consistent with previous studies, with no new safety signals identified.
In addition, the results of two indirect comparison analyses based on the Phase III VALIANT study (EMPAVELI) and the APPEAR-C3G study (iptacopan) showed that EMPAVELI was superior to iptacopan in reducing proteinuria and achieving composite renal endpoints, with more patients reaching a ≥50% reduction in proteinuria and a UPCR below 1 g/g.
This result further validates the long-term efficacy and broad applicability of EMPAVELI in C3G and IC-MPGN. As the only currently approved drug for treating C3G and IC-MPGN patients aged 12 years and older, EMPAVELI is expected to significantly improve the quality of life for patients with these rare kidney diseases.
Empaveli (Pegcetacoplan) is an intravitreally injected, PEGylated cyclic peptide therapy targeting the C3 complement protein developed by the company. It specifically binds to complement C3 and its activation fragment C3b, regulating the overactivation of the complement cascade, thereby inhibiting intra- and extravascular hemolysis as well as inflammatory damage.
Terns Pharma
California, USA, 2025-10-21 — Terns Pharmaceuticals (NASDAQ: TERN) announced the 12-week top-line results of the Phase II clinical trial for its oral GLP-1 receptor agonist TERN-601 in treating obesity. The results showed a maximum placebo-adjusted weight loss of 4.6%, with a discontinuation rate due to adverse events at 12%.
During the follow-up period after treatment, three subjects experienced asymptomatic and reversible Grade 3 liver enzyme elevation events, two of which were considered drug-related. The company stated that this result did not meet the differentiated standards for safety, tolerability, and efficacy expected of oral GLP-1RA therapy, and therefore, it will no longer advance TERN-601 or other metabolism-related projects.
The CEO of the company stated, "Although TERN-601 did not meet expectations, the team will continue to focus on advancing TERN-701, a potential best-in-class allosteric BCR-ABL inhibitor for the treatment of chronic myeloid leukemia (CML). We look forward to announcing new data from its Phase I CARDINAL trial this quarter."
According to the trial data, TERN-601 demonstrated statistically significant weight loss across different dose groups (250 mg to 750 mg), with the 500 mg group achieving the highest placebo-adjusted weight loss of 4.6%. The main adverse reactions were mild to moderate gastrointestinal events, including nausea (56%), vomiting (27%), and constipation (12%), with no severe gastrointestinal events reported.
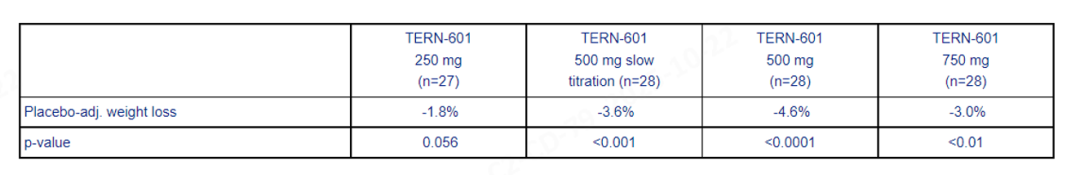
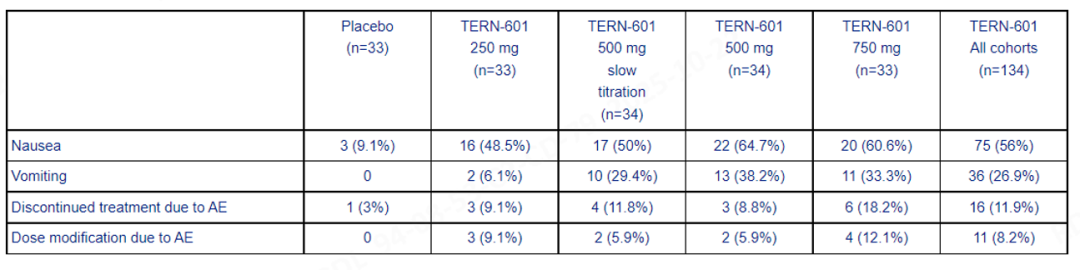
Terns Pharmaceuticals is a clinical-stage biopharmaceutical company focused on developing small molecule drugs to treat major diseases, with its core pipeline currently including products in the oncology and obesity fields.
Hainan Zhonghe Pharmaceutical Co., Ltd.
On 2025-10-22, Hainan Zhonghe Pharmaceutical's semaglutide injection was registered for a clinical trial (CTR20254198) in the CTR. This is a Phase III clinical trial of semaglutide injection for the treatment of obesity.
HaoSen Pharma
On 2025-10-22, Hansoh Pharmaceutical registered a clinical trial (CTR20254127) on the CTR for its GLP-1R agonist HS-10501. This is a multicenter, randomized Phase II clinical study to evaluate the efficacy and safety of once-daily oral HS-10501/HS-10501-2 compared to placebo in subjects with type 2 diabetes.
Registration Approved
Qilu Pharmaceutical
On 2025-10-16, the linaclotide API (registration number: YY20250001041) from Hainan Shuangcheng Pharmaceutical was registered in the CDE’s excipient and packaging material registration system with a status of Inactive.
Enterprise Development
Kailera Therapeutics
United States, October 14, 2025 – Kailera Therapeutics, a clinical-stage biopharmaceutical company, announced the completion of a $600 million Series B financing round led by new investor Bain Capital Private Equity. Other new and existing investors, including Adage Capital, Canada Pension Plan Investment Board (CPP Investments), and Janus Henderson, also participated in the round. The proceeds will be used to advance the global clinical development of the company’s differentiated, late-stage obesity treatment pipeline. Following the completion of this financing round, Andrew Kaplan, a partner at Bain Capital, will join the Kailera board of directors.
Funds Focus on Supporting the Global Phase III Clinical Plan for the Company’s Core Candidate Drug, KAI-9531. KAI-9531 is an injectable GLP-1/GIP dual receptor agonist that has demonstrated potentially best-in-class weight loss effects in clinical trials in China. The phase III plan is expected to launch by the end of this year, covering adult patients with obesity or overweight, including those with or without type 2 diabetes, as well as individuals with a BMI ≥35.
In addition, the financing will also drive the oral small molecule GLP-1 receptor agonist KAI-7535 into global clinical trials and support the development of early projects KAI-4729 (GLP-1/GIP/glucagon triple receptor agonist) and KAI-9531 in oral tablet form. The company also holds the rights to new formulations of certain authorized products and the right of first refusal for Hengrui's metabolic disease portfolio.
Some images and texts are sourced from the internet. If there is any infringement, please contact us for removal.

Flagship Report


Column Recommendation





Peptide Research Society
Biopharmaceuticals · Beauty & Personal Care · Nutrition & Health
Animal Health · Green Agriculture · Biomaterials
Professional Focus | Achieving Customer Success | Growing Together