BioMap and Westlake University Enable Proteins to Perform Computation with Single-Molecule AND Gate

BioMap
Developer of Innovative Drug R&D Platform

In the field of life sciences, cells are often compared to sophisticated miniature computers, constantly processing complex biological signals. For a long time, synthetic biologists have been committed to simulating this computational function by constructing intricate "genetic circuits" or extensive protein networks. However, such "network-level" solutions often face challenges like "bulky devices" (high genetic load), "transport difficulties" (hard to deliver into cells), and "complicated debugging" (difficult to optimize stoichiometric ratios of components).。
The team of Chen Zibo from West Lake University collaborated with BioMap and recently published an article titled "Multivalency Enable" in the journal JACS.s Signal Processing at Single Protein Level" research paper. This study proposes a highly forward-thinking new approach: for simple computational needs, we may not require complex networks but instead canCompressing Signal Processing Functions into a Single Molecule。
By leveraging the multivalency of proteins, the research team successfully endowed individual protein molecules with computer-like logical judgment capabilities, enabling them to precisely sense the identity and density of antigens on the cell surface. This not only significantly reduced the system's complexity but also provided a novel theoretical framework and design tools for developing next-generation "smart drugs" capable of accurately identifying tumor cells.
When proteins possess "logical judgment" capabilities, drugs are no longer just active molecules but begin to have decision-making abilities. BioMap is bringing this cutting-edge technology into real R&D scenarios: relying on affinity models and the AND GATE algorithm, it has designed and validated over 100 bispecific antibody configurations, successfully delivering results.Novel bispecific antibody molecules with a better therapeutic window and stronger selectivity。
At the same time, BioMap's large model system is also continuously evolving, moving from single-point optimization towards a systematic design of orchestratable molecular behaviors, covering300+SOTA Task ModelsAnd form a complete experimental closed loop, providing a high-certainty path for intelligent drug research and development. With the in-depth application of such models in antibody engineering and precision medicine, a safer, more efficient, and predictable future medical system is taking shape at an accelerated pace.
In the field of life sciences, cells are often compared to sophisticated miniature computers, constantly processing complex biological signals. For a long time, synthetic biologists have been committed to simulating this computational function by constructing intricate "genetic circuits" or extensive protein networks. However, such "network-level" solutions often face challenges like "bulky devices" (high genetic load), "transport difficulties" (hard to deliver into cells), and "complicated debugging" (difficult to optimize stoichiometric ratios of components).。
The team of Chen Zibo from West Lake University collaborated with BioMap and recently published an article titled "Multivalency Enable" in the journal JACS.s Signal Processing at Single Protein Level" research paper. This study proposes a highly forward-thinking new approach: for simple computational needs, we may not require complex networks but instead canCompressing Signal Processing Functions into a Single Molecule。
By leveraging the multivalency of proteins, the research team successfully endowed individual protein molecules with computer-like logical judgment capabilities, enabling them to precisely sense the identity and density of antigens on the cell surface. This not only significantly reduced the system's complexity but also provided a novel theoretical framework and design tools for developing next-generation "smart drugs" capable of accurately identifying tumor cells.
When proteins possess "logical judgment" capabilities, drugs are no longer just active molecules but begin to have decision-making abilities. BioMap is bringing this cutting-edge technology into real R&D scenarios: relying on affinity models and the AND GATE algorithm, it has designed and validated over 100 bispecific antibody configurations, successfully delivering results.Novel bispecific antibody molecules with a better therapeutic window and stronger selectivity。
At the same time, BioMap's large model system is also continuously evolving, moving from single-point optimization towards a systematic design of orchestratable molecular behaviors, covering300+SOTA Task ModelsAnd form a complete experimental closed loop, providing a high-certainty path for intelligent drug research and development. With the in-depth application of such models in antibody engineering and precision medicine, a safer, more efficient, and predictable future medical system is taking shape at an accelerated pace.
OneCore Challenge: How to design proteins with "decision-making capabilities"?
1. Limitations of the Design Method:Existing single-molecule control methods (such as allosteric regulation) are extremely complex in design and cannot be modularly assembled like Lego bricks;
2. "Multi-hand" Combined Mathematical Challenge:Multivalent interactions (i.e., a protein extending multiple "hands" to grasp receptors on the cell surface) can enhance binding strength, but the binding process on the cell surface is a highly dynamic non-equilibrium process. Existing thermodynamic models are "too static" to capture dynamic changes, while traditional kinetic models experience exponential computational explosion as the number of protein "hands" increases, making them difficult to use for rapid design.
The core scientific question addressed in this study is precisely: How to establish a biophysical model that is both efficient and precise to guide the design of these "multi-hand" proteins, enabling them to perform accurate Boolean logic operations (such as "AND gates") on the cell surface and distinguish antigen densities?
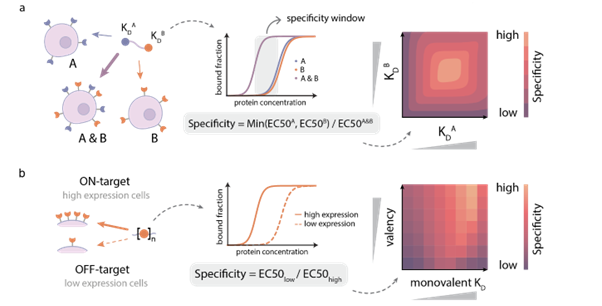
Figure 1: Principles of single-molecule signal processing. (a) Recognition of antigen identity through multivalent binding (e.g., simultaneous recognition of A and B); (b) Sensing of antigen density by adjusting valency and affinity (distinguishing between high-expression and low-expression cells).
2. Research Method: MASS Simulator - A Simulator for Protein Design
In order to solve this problem, the research team developed a set ofMASS (Multivalent Antigen Sensing Simulator)Calculation tool.
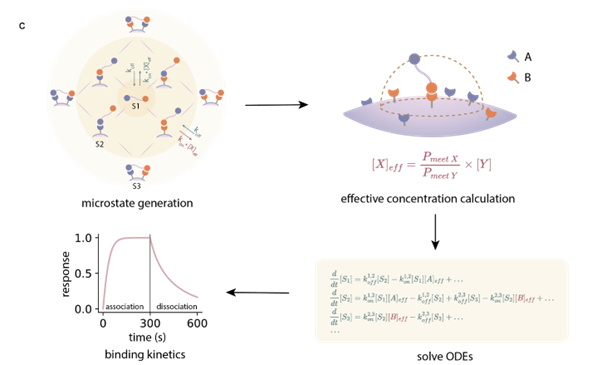
Figure 2: Workflow of MASS. The model deconstructs the complex multivalent binding process into a microstate network, utilizing an innovative algorithm to rapidly calculate effective concentrations, thereby accurately predicting binding kinetics on the cell surface.
MASS is a simulator designed for proteins, with functions including:
Precision Replication:It is based on a kinetic model, breaking down the binding process of proteins to cell surfaces into microscopic states (Microstates), and describing the transitions between them using a system of ordinary differential equations (ODEs).
Algorithm Innovation:The team innovatively introduced a 3D convolution algorithm to calculate the effective concentration, a breakthrough that enables the model to distinguish between two distinct binding modes—"in solution" and "on the cell surface"—and rapidly simulate the binding process of multivalent (multi-handed) interactions.
Multi-dimensional Validation:To ensure the authenticity of the "simulator," the team used biofilm interference technology (BLI), isothermal titration calorimetry (ITC), surface plasmon resonance (SPR), and cell experiments to comprehensively verify the predictive accuracy of the model.
3. Research Conclusion: Precise "Logic Gate" and Sensitive "Density Ruler"
Based on the MASS simulator, the research team successfully achieved two key functions in the experiment:
1. Implementing "AND Gate" Logic: Setting a "Double Lock" for Cells
If a cell is compared to a room, traditional drugs often require only one key (recognizing one antigen) to enter, which can easily lead to the accidental damage of healthy cells. The research team constructed a "bivalent binder" using nanobodies of EGFR and EpCAM, which is analogous to a "double lock" that requires two keys to be inserted simultaneously to open.
Accurate Identification:The MASS model accurately predicted that,This design can preferentially bind to "double-high" cells that simultaneously express two antigens, exhibiting a significant "AND gate" effect.
Intelligent Killing:In a mixed cell population, the constructed antibody-drug conjugate (ADC) specifically killed tumor cells expressing dual antigens, while sparing single antigen cells.
Extension of Three Inputs:The team even successfully designed a trivalent construct targeting three antigens (EGFR, EpCAM, HER2), achieving more complex logical judgment.
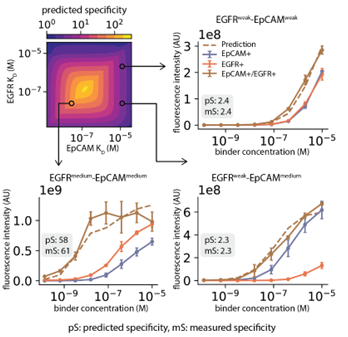
Figure 3: Multivalent constructs achieve cell recognition through logic gating. Demonstrates how bivalent proteins act like logic circuits, binding tightly to cells only when specific antigen combination conditions are met (AND Gate).
2. Antigen Density Sensing: Not Only "Whether or Not", But Also "How Much"
In addition to identifying antigen types, distinguishing antigen density is crucial for differentiating cancer cells (which often highly express specific antigens) from normal cells.
The Wonderful Non-monotonic Relationship:The study found a "non-monotonic" complex relationship between specificity, monovalent affinity, and valency by constructing tandem constructs of PDZ domains with different valencies. Simply put, stronger binding force does not always mean better, and more hands do not always mean better.
Optimal Solution Prediction:The MASS model accurately captured this rule, pointing out that the optimal density resolution capability (Discrimination Power) can be achieved by combining "weak affinity" monomers with a "high-valency" design.
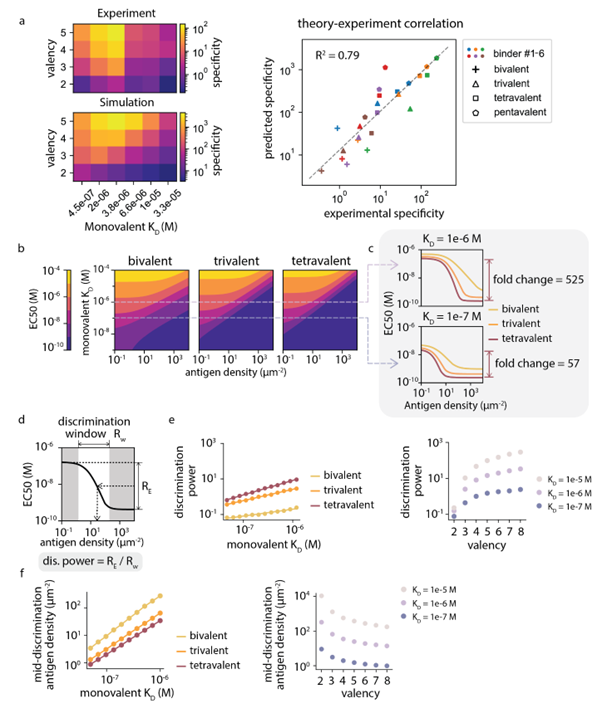
Figure 4: MASS-guided antigen density sensing design. Heatmaps (a, b) demonstrate a high consistency between experiments and simulations; line graphs (c-f) quantify how to precisely adjust the "sensitivity" of proteins to different antigen densities by modulating valency and affinity.
4. Innovation Value: From "Trial and Error" to "Rational Design"
This study is not only a theoretical breakthrough but also a set of implementable engineering solutions.
1. High-efficiency Design Engine:The MASS model breaks through the computational bottleneck, enabling scientists to rapidly screen tens of thousands of design schemes (valence states, structures, affinity combinations) on computers, significantly reducing the "trial and error" costs in the laboratory.
2. Systematic Design Process:The study proposed a complete Pipeline to guide researchers on how to customize super proteins that can both recognize "identity" and sense "density" based on the characteristics of target cells.
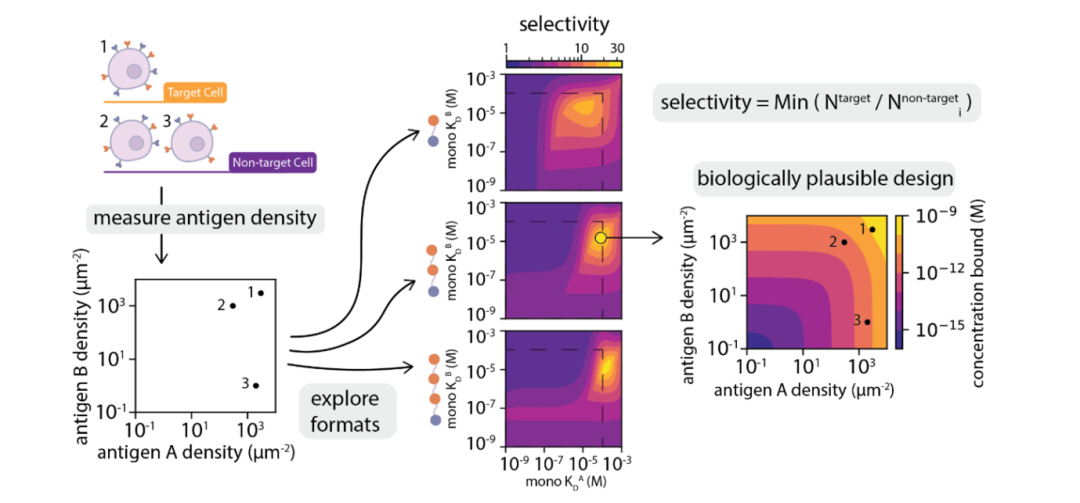
Figure 5: Flowchart of the rational design process for multivalent proteins. Starting from measuring the antigen density of target cells, the optimal combination of valency and affinity is screened through model simulation, ultimately achieving extremely precise recognition of target cells.
V. Conclusion: The Blueprint of the Next Generation of "Smart Drugs"
This work demonstrates that: we can fully utilize biological principles to "encode" complex computational logic into a single protein molecule.
The MASS model is not only applicable to antibody design but also exhibits broad versatility. In the future, as the model further integrates more physical parameters such as temperature and steric hindrance, it is expected to become a standardized design platform in the fields of synthetic biology and biopharmaceuticals. This provides a clear blueprint for the development of next-generation "Smart Drugs" with fewer side effects and more precise efficacy, making the application prospects of "single-molecule computing" in the biomedical field increasingly attainable.
