Over 100 New Drugs Advance in Development; J&J and AstraZeneca Launch First Global Subcutaneous Formulations; GSK’s Twice-Yearly Ultra-Long-Acting Therapy Receives Breakthrough Approval

GSK
Pharmaceutical R&D Manufacturer

According to the "Global New Drugs" module of the Insight database, this week (December 14th - December 20th), a total of 142 innovative drugs (including improved new drugs) worldwide have advanced to new stages of development. Among them, 4 were approved for the first time, 7 filed for marketing authorization for the first time, and 10 registered for Phase III clinical trials for the first time., 30 new Phase I clinical trials registered for the first time.
Below, Insight will introduce the progress of some key projects at home and abroad this week.
*Data Description: The data collection time for this weekly report is: 2025-12-21 13:00. Due to the continuous rapid updates of the Insight database, there may be differences in the timeliness of search results at different times. Please refer to the latest query for accuracy.

On December 16 local time, GSK announced that the U.S. FDA had approvedDermochimab(Depemokimab,Exdensur)Listed asEosinophil PhenotypeSevereAsthmaAdditional Maintenance Medications for Patients, applicable to adult and adolescent patients aged 12 years and above.
The press release pointed out that thisYesThe first and currently only ultra-long-acting biologic approved for the treatment of severe asthma patients with eosinophilic phenotype, with a dosing regimen of twice a year.

Screenshot source: GSK official website
DemochizumabIt is a monoclonal antibody targeting IL-5. This approval is mainly based on SWIFT-1 andSWIFT-2 Two Phase III Clinical TrialsThe positive results.
SWIFT-1 and SWIFT-2 are 52-week, randomized, double-blind, placebo-controlled, multicenter Phase III clinical trials that evaluated the efficacy and safety of Depemokimab as an add-on therapy for patients with eosinophilic asthma.
The results showed that both trials met the primary endpoints. Within 52 weeks, compared with placebo, in the SWIFT-1 and SWIFT-2 studies,Demochizumab respectivelyCan significantly reduce the risk of asthma attacks by 58% in patients with severe asthma.(HR=0.42,95% CI:0.30–0.59,p<0.001)And 48%(HR=0.52,95% CI:0.36–0.73,p<0.001)。
In addition, the trial also achieved secondary endpoints.AcceptDemochizumabThe incidence of exacerbations requiring hospitalization and/or emergency room visits was lower in the treatment group (1% and 4%, respectively) compared to the placebo group (8% and 10%, respectively).
The pre-specified pooled analysis of the two trials showed that, compared with placebo,72% reduction in the risk of clinically significant deterioration requiring hospitalization or emergency treatment(HR=0.28,95% CI:0.13–0.61,p=0.002)。
In these trials,DermochimabWell tolerated, the incidence and severity of side effects in patients were similar to those receiving placebo.
Dermoquimab is GSK's second IL-5 monoclonal antibody to be marketed. The first oneIL-5 Monoclonal AntibodyMepolizumab is the world's first approved IL-5 monoclonal antibody.Since its initial approval in November 2015, sales have increased year by year, reaching $2.279 billion in 2024, a year-on-year increase of 10%.。
On December 15 local time, Daiichi Sankyo and AstraZeneca announced that the U.S. FDA had approvedTrastuzumab Deruxtecan(Enhertu)A new indication has been approved for marketing in China,In combination with pertuzumab for the first-line treatment of adult patients with unresectable or metastatic HER2-positive breast cancerIn November 2025, the marketing application for this indication was also accepted by China's NMPA.

Screenshot source: Daiichi SankyoOfficial Website
Trastuzumab deruxtecan is a DXd ADC targeting HER2 designed using Daiichi Sankyo's proprietary technology. In March 2019, Daiichi Sankyo and AstraZeneca reached a $6.9 billion collaboration to jointly develop and commercialize the product worldwide.
Previously, trastuzumab deruxtecan had received FDA approval for 7 indications, involving HER2-positive breast cancer.(Third-line|Second-line)HER2-Positive Gastric Cancer(Third-line|Second-line), HER2-low breast cancer(Second Line)、HER2-Mutant Non-Small Cell Lung Cancer(Second Line), HER2 low expression or HER2 ultra-low expression breast cancer, etc.
The approval of this new indication is based onGlobal Multicenter Phase III Study DESTINY-Breast09 TrialData were used to evaluate the efficacy of Trastuzumab Deruxtecan with or without Pertuzumab versus standard treatment THP.(Paclitaxel, Trastuzumab, and Pertuzumab)Efficacy and safety as a first-line drug for treating HER2-positive, advanced or metastatic breast cancer patients.The primary efficacy endpoint was based on RECIST v1.1 criteria, as assessed by blinded independent central review.(BICR)Assessed Progression-Free Survival(PFS). The key secondary efficacy endpoint was overall survival(OS). Other secondary endpoints include investigator-assessed PFS, confirmed objective response rate(ORR)And Duration of Relief(DOR)。
DESTINY-Breast09 data were presented at the 2025 ASCO Annual Meeting. In this trial,Median in the THP Group PFS OnlyFor26.9 months, whileMedian PFS reached 40.7 months in the trastuzumab deruxtecan + pertuzumab group, reducing the risk of disease progression or death by 44%.At the same time, compared with the THP group, the PFS benefit of the trastuzumab deruxtecan + pertuzumab group remained consistent across all subgroups.
Trastuzumab Deruxtecan + Pertuzumab GroupConfirm ORR at 87%, while the THP group was 81%. In the trastuzumab deruxtecan + pertuzumab group, 15% of patients achieved complete remission.(CR)And 72% of patients achieved partial remission.(PR), while in the THP group, 8% of patients achieved CR and 73% achieved PR. In the trastuzumab deruxtecan + pertuzumab group,Median DOR exceeds three years(39.2 months), while the THP group is approximately two years.
In the DESTINY-Breast09 study, the safety profile of trastuzumab deruxtecan + pertuzumab was consistent with the known safety profiles of each monotherapy, with no new safety issues identified. Grade 3 or higher treatment-emergent adverse events occurred in 54.9% and 52.4% of patients in the trastuzumab deruxtecan + pertuzumab group and the THP group, respectively.(TEAE)。
In the trastuzumab deruxtecan + pertuzumab treatment group, the most common Grade 3 or higher TEAEs with an incidence rate ≥5% included: neutropenia, hypokalemia, anemia, fatigue, diarrhea, thrombocytopenia, and nausea. According to the assessment by the Independent Adjudication Committee, 12.1% of patients receiving trastuzumab deruxtecan + pertuzumab treatment developed interstitial lung disease. (ILD) Or pneumonia events. Most ILD or pneumonia events are low-grade.(Grade 1 or Grade 2)Two grade 5 ILD events occurred in the trastuzumab deruxtecan + pertuzumab group.
On December 16, AstraZeneca announced that itsSubcutaneous Injection of Biologics Saphnelo (Anifrolumab,Avelumab Subcutaneous Injection)Approved in the EU, can be used asPre-filled injection pen, forSystemic Lupus Erythematosus(SLE)Adult patientsSubcutaneous Self-Administration, as a supplement to standard therapy.
Screenshot source: AstraZeneca official website
Saphnelo isThe World's FirstApproved Targeted Drugs for SLE Treatment Targeting the Type I Interferon Pathway, can interact with Type I interferon(IFN)Receptor subunit 1 binding, thereby blocking the activity of type I interferons.The drug wasFirst FDA approval in July 2021 for the treatment ofAdult patients with moderate to severe SLE, and was successively approved for marketing in Japan and the EU in September 2021 and February 2022. Since its approval,The sales of Avelumab have been continuously increasing.Achievements in the First Three Quarters of 2025$4.83 billion, a year-on-year increase of 47%.
This time isSaphnelo Subcutaneous FormulationsThe World's First Approval. Apart from the EU,Saphnelo Subcutaneous Injection is Under Regulatory Review in Multiple Countries, Including the United States and Japan.
In September this year, AstraZeneca announcedSaphnelo Subcutaneous Formulation Targets SLE Planned Interim Analysis of Phase III TULIP-SC Trial in PatientsAchieve the primary endpoint。
TULIP-SC is aPhase 3, multicenter, multinational, randomized, double-blind, placebo-controlled study designed to evaluate the standard of care (SOC) Patients with moderate to severe active autoantibody-positive SLE treated with subcutaneous injectionSaphnelo Efficacy and safety compared to placebo.
Results show that, compared with placebo, Saphnelo subcutaneous injection therapySignificantly reduced disease activity, and is clinically significant. The safety observed in the interim analysis is consistent with the known clinical profile of Saphnelo intravenous infusion.
On December 17, Johnson & Johnson announced,The U.S. FDA has approved the subcutaneous injection formulation RYBREVANT FASPRO™ of the EGFR/c-MET bispecific antibody "amivantamab" for marketing in the United States, applicable to all indications already approved for the intravenous formulation RYBREVANT® (amivantamab-vmjw).
The press release noted that this is the first and only subcutaneous injection approved for EGFR-mutated NSCLC.

Screenshot source: Corporate official website
Compared with intravenous injection(IV)Compared with traditional administration, RYBREVANT FASPRO™ significantly improves patient convenience and reduces the burden on medical resources:
Reduce the administration time from several hours to 5 minutes;
Administration-related reactions(ARR)The incidence rate decreased by approximately five times.(Subcutaneous injection and intravenous injection were 13% vs 66%, respectively);
Reduced the risk of venous thromboembolism(VTE)The incidence rate (11% vs 18%)
Based on Phase 3 PALOMA-3 Study(NCT05388669)As a result, RYBREVANT FASPRO™ demonstrated consistent efficacy with RYBREVANT®, achieving the two co-primary pharmacokinetic endpoints.(PK)Endpoint. The results of the PALOMA-3 study were first presented as a late-breaking oral report at the 2024 American Society of Clinical Oncology (ASCO).(ASCO)Announced at the annual meeting and published in the Journal of Clinical Oncology.
The results also showed that, compared with intravenous injection(IV)Compared to the group, subcutaneous injection(SC)Duration of Remission in the Group(DoR)Longer, Progression-Free Survival(PFS)Improved, Overall Survival(OS)Also longer. The median overall survival in patients treated with subcutaneous injection combined with LAZCLUZE® was significantly longer.(Hazard Ratio [HR] 0.62; 95% Confidence Interval [CI] 0.42 - 0.92; nominal P value = 0.02)At 12 months, 65% of patients treated with subcutaneous injections were alive, compared to 51% of those treated with intravenous infusions.
On December 19 local time, Cytokinetics announced that the U.S. FDA had approved the marketing application for its next-generation cardiac myosin inhibitor, Aficamten, for the treatment of symptomaticObstructive Hypertrophic Cardiomyopathy(oHCM)Adult patients. The rights to the drug in the Greater China region belong to Sanofi, and on December 17,Approved in China。

ComeSource:Cytokinetics Official Website
Aficamten is a selectiveSmall Molecule Cardiac Myosin InhibitorsIn preclinical models, Aficamten prevents this by directly binding to cardiac myosin at a unique, selective allosteric binding site.Myosin enters the force-producing state to reduce myocardial contractility.
This approval is based on the results of the pivotal Phase III clinical study SEQUOIA-HCM. The results of SEQUOIA-HCM indicate that, compared with placebo,Aficamten Treatment for 24 Weeks Significantly Improves Exercise Capacity, compared with baseline, cardiopulmonary exercise test in patients treated with Aficamten(CPET)The measured peak oxygen uptake(pVO2)Increased by 1.8 ml/kg/min, while the placebo-treated patients showed 0.0 ml/kg/min, with a least squares mean difference of 1.74. (1.04 - 2.44) mL/kg/min(95% CI)(p=0.000002)。Aficamten The treatment effect remained consistent across all predefined subgroups.
In July 2020, Jixing Pharmaceuticals and Cytokinetics reached a collaboration, obtaining the exclusive license for the development and commercialization of Aficamten in Greater China. In December 2024,Sanofi partners with Jixing Pharmaceuticals to obtain the rights in Greater China for the drug.

Orforglipron is an investigational, once-daily oral small molecule(Non-peptide)Glucagon-Like Peptide-1 Receptor Agonist(GLP-1 RA). The medication can be taken at any time of the day without restrictions on food or water intake. It has already met the primary endpoints in multiple Phase III studies.
Analysts at Truist Securities predict that Orforglipron has a huge market opportunity, with expected global peak sales reaching$14.7 billion。
In this study, for participants in the SURMOUNT-5 trial who had previously reached a weight plateau, Orforglipron achieved the primary endpoint of maintaining a higher percentage of weight loss compared to placebo. In the pre-specified 52-week analysis,Participants Switching from Wegovy to Orforglipron Maintained Prior Weight Loss, with an average difference of 0.9 kg; andParticipants switching from Zepbound to Orforglipron maintained their previously achieved weight loss., with an average difference of 5.0 kg(Using efficacy estimates)。
In the post-hoc analysis, at 24 weeks(Last time point before participants in the placebo group met the criteria for Orforglipron as a rescue treatment)Patients switching from Wegovy to Orforglipron experienced a weight change of -0.1 kg from the ATTAIN-MAINTAIN baseline, compared to 9.4 kg in the placebo group. Similarly, patients switching from Zepbound to Orforglipron had a weight change of 2.6 kg from baseline, compared to 9.1 kg in the placebo group.

The new drug application is based on the results of two studies: REDEFINE 1 and REDEFINE 2.
REDEFINE 1 is a 68-week Phase III randomized, double-blind, placebo- and active-controlled trial designed to evaluate once-weekly CagriSema compared with 2.4 mg alone.Cagrilintide, the efficacy and safety of using 2.4 mg semaglutide or placebo alone compared to lifestyle intervention as an adjunctive therapy. All participants were 3,417 obese individuals.(BMI ≥ 30 kg/m²)Or overweight(BMI ≥ 27 kg/m² )And accompanied by one or more obesity-related complications but without diabetes in adults.
REDEFINE 2 is a 68-week Phase III double-blind, randomized, placebo-controlled trial designed to evaluate the efficacy and safety of once-weekly CagriSema compared to placebo, as an adjunct to lifestyle intervention. The trial enrolled 1,206 adults with type 2 diabetes who were also obese or overweight.
REDEFINE 1 trial found that, regardless of whether patients adhered to the treatment, those receiving CagriSema showedWeight loss of 20.4% at 68 weeks(Average baseline weight was 236 pounds), while the placebo group only reduced by 3.0%(Average baseline weight was 235 pounds), the difference was statistically significant.
If all patients adhere to the treatment, in the evaluation of treatment efficacy, the CagriSema groupGreater weight loss of 22.7% at Week 68, while the placebo group was only 2.3%.
In subjects taking CagriSema,91.9% of weight loss reached or exceeded 5%, while the proportion in the placebo group was 31.5%.
In addition, a supportive secondary analysis showed that about half of the trial participants who were obese at baseline.(54%)Reached non-obesity standards at Week 68 after receiving CagriSema treatment(BMI < 30 kg/m² )In the placebo group, only 11.1% of participants met the criterion at week 68.
In terms of safety, the safety data generated in the REDEFINE 1 and 2 trials were comparable to those of GLP-1 receptor agonists.
The US FDA is expected to review the application for CagriSema in 2026.

Source:ClinicalTrials Official Website
This isA Phase III randomized, double-blind, placebo-controlled study,Proposed to include 1,035 people,The main purpose is to evaluate, compared with placebo,Once-weekly injection Eloralintide Efficacy and Safety of Weight Loss in Subjects Who Are Overweight or Obese and Have Type 2 Diabetes. The study will last approximately 75 weeks.
Eloralintide is an investigational once-weekly, selective amylin receptor agonist. In 2025At the annual Obesity Week conference, Eli Lilly announced positive results from a Phase II clinical trial of the drug.
This is a 48-week randomized, double-blind, placebo-controlled trial evaluating the efficacy and safety of once-weekly Eloralintide monotherapy compared to placebo in obese or overweight adult participants without type 2 diabetes but with at least one weight-related comorbidity.The primary objective of the study is to demonstrate the superiority of Eloralintide over placebo in terms of the percentage change in body weight from baseline at Week 48.。
The study randomly enrolled 263 participants in the United States, allocating them in a 2:1:1:1:2:1:2 ratio to the following groups: placebo, Eloralintide 1 mg, 3 mg, 6 mg, 9 mg, or escalating dosing regimens of 6/9 mg and 3/6/9 mg respectively.
The results showed,At Week 48, all dose groups of Eloralintide reached the primary endpoint,The average weight loss ranged from 9.5% to 20.1%., surpassing the average reduction of 0.4% in the placebo group(Use Effectiveness Estimation Target)In addition, all dose groups of Eloralintide showed weight reduction and BMI improvement, etc.Showed clinically meaningful improvement over placebo in secondary endpointsEloralintide is also associated with improvements in multiple cardiovascular risk factors, including waist circumference, blood pressure, blood lipids, blood glucose control, and inflammatory markers.
On December 15, Sanofi announced the PERSEUS Phase III clinical study(NCT04458051)The results showed,BTK Inhibitor Tolebrutinib Fails to Meet Primary Endpoint, i.e., failed to delayPrimary Progressive Multiple Sclerosis(PPMS)Patient6-Month Confirmed Disability Progression(cCDP)Time.

Screenshot source: Sanofi official website
Tolebrutinib is a brain-penetrant and bioactive BTK inhibitor, with its core advantage being the precise targeting of multiple sclerosis.(MS)The Key Factor Driving Disability Progression —— Neuroinflammation. Unlike existing MS therapies that primarily target peripheral inflammation, Tolebrutinib can penetrate the blood-brain barrier and reach therapeutic concentrations in cerebrospinal fluid, thereby regulating B lymphocytes within the central nervous system and disease-associated microglia.
PERSEUS is a global, double-blind, randomized Phase III clinical trial designed to evaluate the efficacy and safety of Tolebrutinib compared to placebo in patients with PPMS. Participants were randomly assigned.(2:1)Participants will receive either daily oral Tolebrutinib or a matching placebo for a treatment period of up to approximately 60 months. The primary endpoint of the study is six-month confirmed disability progression.(cCDP)Time of occurrence.
Based on the results of the PERSEUS Phase III clinical trial, Sanofi stated that it will not seek regulatory approval for Tolebrutinib in the treatment of PPMS.
However, preliminary analysis shows that the safety profile of Tolebrutinib is consistent with previous study results. As previously reported, drug-induced liver injury(DILI)Is a known risk of Tolebrutinib. Strict adherence to liver function monitoring requirements and timely management of elevated liver enzymes are crucial for reducing the risk of DILI. Complete safety and efficacy results will be presented at an upcoming medical conference.
Autoimmunity has become a new battleground for BTK, with Sanofi taking the lead in this field. In August this year, the company's first...BTK Inhibitor RilzabrutinibApproved in the United States for the treatment of immune thrombocytopenia, becoming the world's first BTK inhibitor specifically developed for autoimmune diseases.
Although the second BTK inhibitor, Tolebrutinib, stumbled in the Phase III trial for PPMS, itsNon-relapsing Secondary Progressive Multiple Sclerosis(SPMS)The indications have received interim approval in the United Arab Emirates and are currently undergoing regulatory review for market application in the European Union, the United States, and other countries and regions.. In the United States, Tolebrutinib targetsSPMSThe PDUFA date for the indication is December 28.
Pharmaceutical Transactions
According to the Insight database, a total of 53 transaction events occurred this week (December 14 - December 20).
On December 15, Fosun Pharma announced that its holding subsidiary, Fosun Pharmaceutical Industry, plans to invest a total of 1.41248 billion yuan to take a controlling stake in Green Valley Pharma. This move aims to further enrich Fosun Pharma's innovative product pipeline matrix in the field of central nervous system treatment, improve market layout, and create an integrated diagnosis-treatment solution with multi-technology pathway synergy.

Source of screenshot: Fosun Pharma announcement
Without considering other factors that may affect the equity structure changes of the target company, upon the completion of this acquisition, Fosun Pharma will hold 53% of LuGu Pharma’s equity through Fosun Pharma Industry and SPV. LuGu Pharma will be included in the scope of Fosun Pharma's consolidated financial statements as a subsidiary. If the subsequent transfer is completed, Fosun Pharma will hold 51% of LuGu Pharma’s equity through Fosun Pharma Industry.
Green Valley Pharmaceuticals was established in October 2018, primarily engaged in the research, development, production, and sales of treatments for neurodegenerative diseases. In November 2019, Green Valley's sodium oligomannate capsule received conditional approval from the National Medical Products Administration (NMPA) for marketing, with the approved indication being "for mild to moderate Alzheimer's disease to improve patients' cognitive function." In 2021, the drug was included in China’s national medical insurance catalog.
Due to the expiration of the registration certificate, the commercial production of Ganlute Sodium Capsules has not been carried out since November 2024. Before the commercial production and sales of this drug can resume, it is required to (mainly including) complete the post-marketing confirmatory clinical trial and obtain approval from the national drug evaluation authorities. As of the date of this announcement, the post-marketing confirmatory clinical trial is already underway.
On December 16, Hansoh Pharma announced that it had entered into an agreement with Glenmark Specialty S.A. regarding Ameletinib.Exclusive License, Collaboration and Distribution Agreement。

Source of screenshot: Corporate announcement
According to the license agreement, Hansoh Pharma will grant Glenmark an exclusive license, allowing it to operate in the authorized region.(Middle East and Africa, Southeast Asia and South Asia, Australia, New Zealand, Russia and other CIS countries, as well as certain specific Caribbean countries covered by the agreement)Develop and commercialize Ameitinib.
In return, Hansoh Pharma will receive an upfront payment and potential subsequent cumulative payments.More than one billion US dollarsRegulatory and commercial milestone payments, as well as tiered royalties on net sales within the licensed territory.
Ametinib isChina's First Original Third-Generation EGFR-TKI Innovative Drug,Has been approved for four indications in China.June 2025, Ameitinib(UK product name Aumseqa®)Awarded by the UK Medicines and Healthcare products Regulatory Agency(MHRA)Approved for marketing.
On December 17, Harbour BioMed announced,Entered into a Long-term Global Strategic Collaboration and License Agreement with Bristol-Myers SquibbThe two parties will jointly develop a new generation of multispecific antibody therapies.
Source: HBM official WeChat
According to the terms of the agreement, Harbour BioMed will collaborate with Bristol-Myers Squibb to advance and accelerate the multispecific antibody discovery program. Harbour BioMed will receiveTotal payment of 90 million US dollars,If Bristol-Myers Squibb chooses to advance all potential projects, HBM BioPharma could receive up to$1.035 billion for development and commercializationMilestone Payment, as well as tiered royalties based on future product net sales.
December 15th evening,Changchun High-TechThe holding subsidiary, Jin Sai Pharmaceutical, authorizes its wholly-owned subsidiary, Sai Zeng Healthcare,As the technology licensor,Exclusive License Agreement Signed with Yarrow Bioscience, Inc. for GenSci098 Injection Project.

Screenshot source: Corporate announcement
According to the terms of the agreement, Yarrow will obtain GenSci098 Injection outside Greater China.(Mainland China, Hong Kong Special Administrative Region, Macao Special Administrative Region, and Taiwan Region)Global exclusive rights for development, production, and commercialization outside of China, for GenSci098 injection specifically targeting thyroid-associated ophthalmopathy.(TED)and Diffuse Toxic Goiter(GD)Research, development, and commercialization of indications such as...
Saizen Medical retains the development and commercialization rights for GenSci098 Injection in China. In the future, the intellectual property developed by each party will belong to each respective party, but they will grant each other licenses and authorization to use such intellectual property.
In return, Saizen Medical is expected to receive$120 millionDown Payment and Recent Development Milestone Payments(Including a non-refundable, non-creditable upfront payment of 70 million US dollars and subsequent near-term development milestone payments of 50 million US dollars), and will be eligible to receive milestone payments related to specific R&D, regulatory, and commercialization milestones. Saizen Medical will have the right to obtain up to [amount] for this exclusive license.$13.65 billionMilestone payments, with the right to receive over net sales after subsequent product launch10% sales commission.
GenSci098 Injection is a humanized thyroid-stimulating hormone receptor developed independently by Jin Sai Pharmaceutical.(TSHR)Antagonistic monoclonal antibody, classified as a Class 1 new biologic for therapeutic use.Currently, GenSci098 Injection is used for the treatment ofThyroid-associated ophthalmopathy(TED)AndDiffuse Toxic Goiter(GD)Relevant Indications Research.
ADEL-Y01 is a humanized monoclonal antibody,Optionally targets the acetylation of lysine at position 280 Tau Protein (acK280). Unlike therapies targeting total tau protein, ADEL-Y01Specifically inhibits the aggregation and spread of toxic tau proteins(This is a key driver of Alzheimer's disease pathology), while preserving the function of normal microtubule-associated tau protein. Currently, ADEL-Y01 is undergoing a global Phase I clinical trial.
The strategic cooperation with Dren Bio aims to jointly discover and developNext-Generation B Cell Depletion Therapy for the Treatment of Various Autoimmune DiseasesAccording to the terms of the agreement, Dren Bio will receive$100 millionThe down payment, and are eligible to receive up to$1.7 billionDevelopment, regulatory and commercial milestone payments.
The two parties will collaborate on drug discovery and preclinical development activities using Dren Bio's proprietary platform. After selecting the candidate drug, Sanofi will take charge of subsequent development, manufacturing, regulatory, and commercialization efforts.
Dren Bio can opt for a U.S. profit/loss sharing agreement with Sanofi. If this agreement is chosen, Dren Bio willJointly bear 40% of the global ongoing development costs, in exchange for co-promotion rights in the United States and a share of profits/losses in the U.S. region.50/50 splitDren Bio is still eligible for milestone payments and tiered royalties on net sales outside the United States.
It is worth mentioning that in March this year, the two parties have already reached an agreement on the myeloid cell bispecific antibody DR-0201.Acquisition Agreement, The total transaction amount is$19 Billion, including a down payment of 600 million US dollars.

December 17, Sanofi announced,"Ah FuKaitai」(Aficamten, Trade Name: Xing Shu PingApproved for marketing in China, for the treatment ofNew York Heart Association(NYHA)Heart Function Class II-III Obstructive Hypertrophic Cardiomyopathy (HCM)Adult patients, to improve motor capacity and symptoms。
Previously, the drug received breakthrough therapy designations in both China and the U.S. Its marketing application in China was also included in the priority review category, allowing for rapid approval.. This week, the drug has also been approved for marketing by the U.S. FDA.
Aficamten(Research Code: CK-274)YesA New Generation of Selective Small Molecule Cardiac Myosin Inhibitors, mostAs early asDeveloped by Cytokinetics. In July 2020, Jixing Pharmaceuticals reached a collaboration with Cytokinetics and obtained the rights for Greater China. In November 2024, Bayer acquired the rights for Japan.Sanofi reached a cooperation with Jixing Pharmaceuticals in December of the same year, obtaining the rights to the drug in the Greater China region.
The approval of Aficamten is based onKey Positive Topline Results from Phase 3 Registrational Clinical Trial SEQUOIA-HCM.This is aRandomized、Double-blind、PlaceboIn comparison to the international multicenter Phase 3 clinical trial, a total of 282 patients were enrolled.Randomly assigned to the Aficamten group(Starting dose 5mg, maximum dose 20mg)Or placebo group, treatment lasted for 24 weeks with dose adjustments based on echocardiography results.
In terms of safety, in the SEQUOIA-HCM China subgroup, Aficamten was well-tolerated, with adverse events overall comparable to placebo.
On December 15, the CDE website showed that Qilu Pharmaceutical's Class 2.3 new drugAprepitant Palonosetron Emulsion for InjectionThe marketing application has been accepted. Based on the progress of the clinical research of this drug, the Insight database speculates that the indication for this application is forPrevention of Nausea and Vomiting Caused by High-Risk Emetogenic Chemotherapy Regimens。
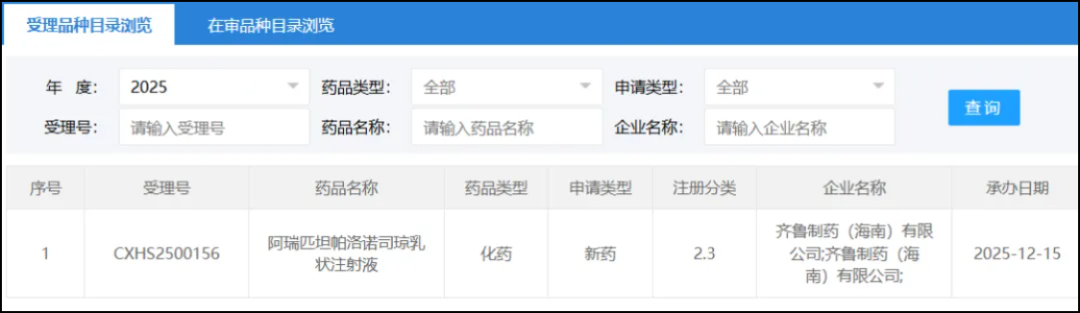
Screenshot source: CDE official website
Aprepitant, originally developed by Merck, is a neurokinin-1(NK-1)Receptor antagonist, mainly used for the prevention and treatment of nausea and vomiting caused by chemotherapy or surgery. In China, in addition to the original drug, there are also multiple generic versions of Aprepitant on the market. Among them, developed by Qilu PharmaceuticalAprepitant InjectionAndAprepitant CapsulesBoth formulations have been approved.
Palonosetron is a 5-HT3 receptor antagonist, mainly used for the prevention and treatment of nausea and vomiting caused by chemotherapy or surgery. The original research drug was developed by Helsinn Group in Switzerland.(Helsinn)R&D. In October 2021, Fosun Pharma reached an agreement with Helsinn Group to obtain products including Netupitant and Palonosetron Capsules.(Product name: Oconzee)The exclusive licensing, distribution, and promotion rights for multiple related tumor supportive care drugs, including in mainland China, Hong Kong, and Macao. Among them, the Oconzee capsule was approved for marketing in mainland China in 2019.
In China, in addition to the original Palonosetron drug, there are multiple generic drugs already on the market, including the Palonosetron Hydrochloride Injection developed by Qilu.
Qilu Pharmaceutical's submission this time isAripitant and Palonosetron Emulsion for Injection(Development Code QLM2010)。
According to the Insight database, in October this year, Qilu has completed a multi-center, randomized, double-blind, positive drug parallel-controlled Phase III clinical study in China.(CTR20242556), this trial aims to evaluate the efficacy and safety of QLM2010 in preventing nausea and vomiting induced by highly emetogenic chemotherapy regimens.
On December 18, Bio-Thera Solutions announced its Class 1 new drugVilaxitamab(BAT5906)The marketing application has been accepted by China's NMPA for proposed use inTreatment of Neovascularization(Wet) Age-related Macular Degeneration(nAMD)。BAT5906 It is a recombinant anti-VEGF humanized monoclonal antibody. InSight database shows that this isThe Second New Drug of Bio-Thera to be Marketed.

Screenshot source:Bio-Thera Announcement
BAT5906 is an innovative recombinant humanized monoclonal antibody drug independently developed and produced by Bio-Thera, with a full-length IgG1 antibody structure and a molecular weight of 149 kDa. It specifically binds to human VEGF-A165 and inhibits new blood vessel formation.
In an in vitro angiogenesis model, BAT5906 was able to block VEGF from binding to its corresponding receptor, inhibiting endothelial cell proliferation and new blood vessel formation.
In animal experiments, BAT5906 has a longer half-life in monkey vitreous than the Fab fragment-based ranibizumab, which may support a longer injection interval in clinical settings. In terms of safety, it does not trigger antibody-dependent cell-mediated cytotoxicity. (ADCC), thus causing fewer systemic adverse reactions, and its clinical application may be safer.
Currently, BAT5906 is targetingDiabetic Macular Edema(DME)Phase III clinical trials for the indications are currently underway; targetingMacular Edema Secondary to Central Retinal Vein Occlusion(CRVO-ME)Indications and TargetingChoroidal Neovascularization in Pathological Myopia(pmCNV)Phase II/III clinical studies for the indication are currently underway.
On December 16, the CDE website showed that Hengrui had filed a Class 1 new drug.SHR-A1904 for InjectionProposed for inclusion in the breakthrough therapy category, with the indication beingPreviously treated with at least one line of systemic therapy for locally advanced or metastatic gastric or gastroesophageal junction adenocarcinoma positive for CLDN18.2(GC/GEJC)。
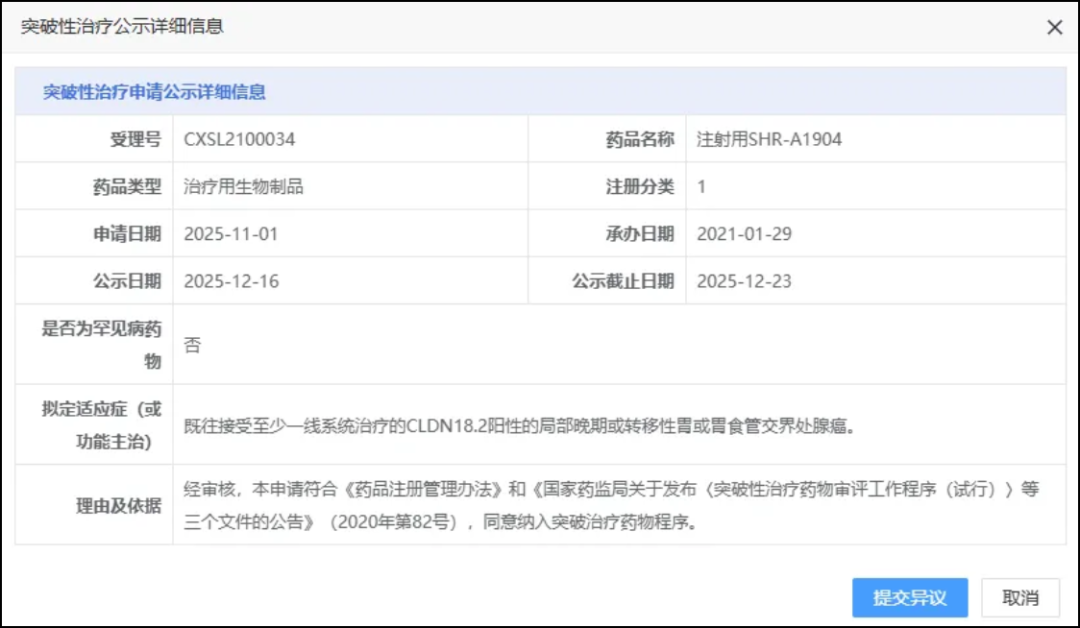
SHR-A1904 is a self-developed and intellectual property-owned antibody-drug conjugate targeting Claudin 18.2 by Hengrui.(ADC), whose payload is a topoisomerase inhibitor(TOPOi),By binding to the target antigen on the surface of tumor cells, the drug is internalized into the cells and releases small-molecule toxins to kill tumor cells.
In October 2023, Hengrui will launch the SHR-A1904 projectLicensed to Merck for a feeThe latter will obtain: 1) the exclusive option to develop, manufacture, and commercialize SHR-A1904 globally outside of mainland China; 2) the option to co-commercialize SHR-A1904 in mainland China with Hengrui.
July 2025Month, HengruiAnnouncementSHR-A1904Phase I Clinical Study in Patients with Advanced Solid Tumors Published in"Nature Medicine." TheThe study enrolled a total of 95 patients with CLDN18.2-positive advanced GC/GEJC.
During the study period, patients received 0.6 to 8.0 mg/kg once every three weeks.(Q3W)SHR-A1904 Intravenous Therapy(0.6 mg/kg, n=2; 1.2 mg/kg, n=3; 2.4 mg/kg, n=3; 3.6 mg/kg, n=3; 4.8 mg/kg, n=9; 6.0 mg/kg, n=35; 8.0 mg/kg, n=40)。
During the dose-escalation phase, SHR-A1904 demonstrated good tolerability. The study selected 6.0 mg/kg and 8.0 mg/kg Q3W to continue exploration in the PK expansion and efficacy expansion phases. In terms of safety, the overall safety of SHR-A1904 was manageable in 95 patients.
In terms of efficacy, exploratory biomarker analysis found that the expression level of CLDN18.2 was positively correlated with efficacy, and patients with medium to high expression of CLDN18.2 responded better to SHR-A1904 treatment. Among 74 evaluable patients,One patient achieved complete remission, 25 patients achieved partial remission, with an ORR of 35.1%, and a confirmed ORR of 23.0%.。
In the two dose groups of 6.0 mg/kg and 8.0 mg/kg,The confirmed ORR was 26.7% and 26.5%, respectively.; The median duration of response was not reached and 8.1 months, respectively;Median PFS was 5.6 months in the overall population., at doses of 6.0 mg/kg and 8.0 mg/kg, respectively5.6 months and 5.5 months.
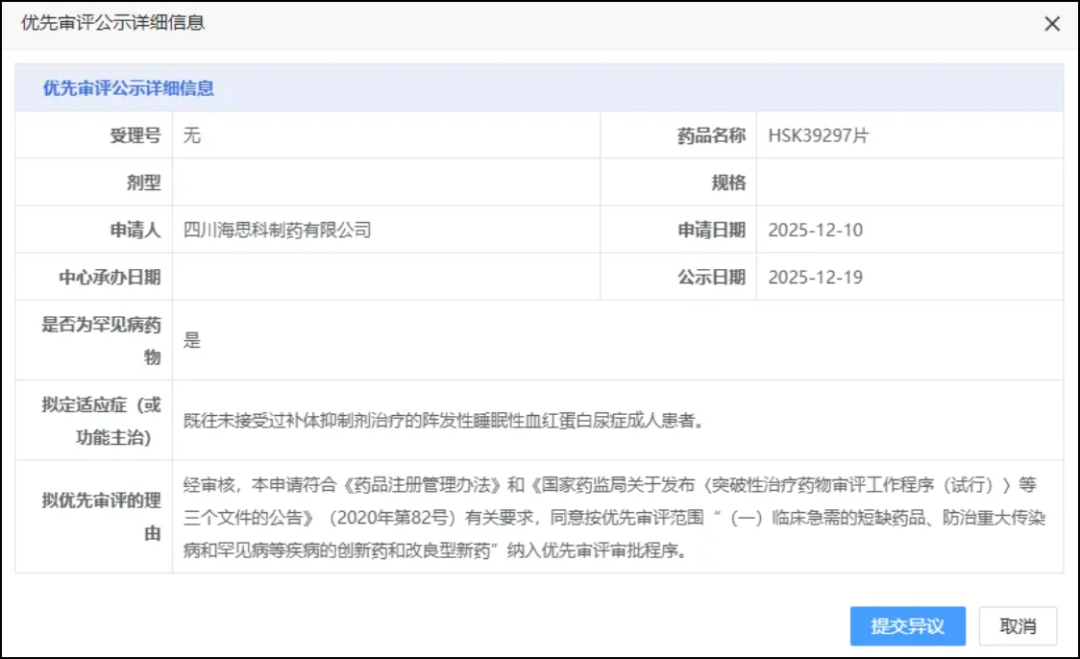
On December 14, InnoCare announced its BTK inhibitorOrelabrutinib in the Treatment of Systemic Lupus Erythematosus(SLE)Reached Primary Endpoint in Phase IIb Clinical Study, while also receiving CDE approval to conduct Phase III pivotal clinical trials.
Screenshot source:Official WeChat Account of InnoCare Pharma
Stage IIbClinical results show that orelabrutinib demonstrated excellent efficacy and good tolerability and safety in patients who received treatment for 48 weeks. This study enrolled a total of 187 patients, who were randomly divided into three groups in a 1:1:1 ratio, namely two dose groups receiving oral orelabrutinib once daily at 75 mg and 50 mg, respectively, and one placebo group.
The primary endpoint of this study is the SLE Responder Index-4 at Week 48.(SRI-4)Response Rate. At Week 48, the SRI-4 response rate in the once-daily 75 mg orelabrutinib group was significantly higher than that in the placebo group.(57.1% vs. 34.4%), statistically significant(p<0.05), reaching the primary endpoint. In addition, the efficacy of the 75 mg orelabrutinib once-daily dose group was superior to that of the 50 mg once-daily dose group, indicating a dose-dependent trend of improvement in efficacy.
At Week 48, the SRI-6 response rate and BICLA composite assessment for the group receiving 75 mg of orelabrutinib once daily(BICLA)The response rate was significantly higher than that of the placebo group, with statistical significance.(p<0.05), achieving the secondary endpoint.
In subgroup patients with baseline disease activity BILAG ≥1A or ≥2B, the SRI-4 response rate in the 75 mg once-daily orelabrutinib group increased by 35% compared to the placebo group. In subgroup patients with baseline disease activity BILAG ≥1A or ≥2B and clinical SLEDAI-2K score ≥4, the SRI-4 response rate in the 75 mg once-daily orelabrutinib group increased by 43% compared to the placebo group.
At the same time, orelabrutinib demonstrated good tolerability and safety, with safety characteristics consistent with the mechanism of action of BTK inhibitors and the disease biology of SLE.
The press release from InnoCare Pharma pointed out that Orelabrutinib is the world's first BTK inhibitor to demonstrate significant efficacy in a Phase II clinical trial for SLE, with its Phase IIa clinical data for the treatment of SLE previously presented at the European Congress of Rheumatology.(EULAR)Released as a重磅 oral report.
The first three quarters of 2025,Orelabrutinib revenue increased by 45.8% year-over-year, reaching 1.01 billion yuanYuan. As new indications continue to expand, Orelabrutinib's future sales are expected to grow steadily.
On December 15, Huadong Medicine announcedCloth,HDM1005 InjectionInfusionPositive results were achieved in the Phase II clinical trial in China for the weight management indication.
Screenshot source: Official WeChat account of the company
This Chinese Phase II clinical trial is a multicenter, randomized, double-blind, placebo-controlled parallel group Phase II clinical study evaluating the efficacy and safety of HDM1005 injection in obese non-diabetic adult subjects. A total of 243 subjects were enrolled, with a dosing period of 22 weeks. Prior to dosing, baseline characteristics such as weight, waist circumference, and BMI were balanced across all dose groups.
The study results showed that, based on the therapy strategy, the weight changes from baseline after 22 weeks of titration every 4 weeks for the HDM1005 injection 0.5 mg group, 1.0 mg group, 2.0 mg group, and 4.0 mg group were respectively-7.47%、-9.73%、-13.31%、-13.28%, -2.46% in the placebo group.
HDM1005 0.5 mg to 4.0 mg dose groupsThe proportion of subjects with a weight reduction ≥10% was 24.0%、52.1%、75.0%、70.8%, while the placebo group was 6.1%.
After 22 weeks of administration, the changes in waist circumference and BMI from baseline for HDM1005 injection were -6.3 to -10.3 cm and -2.4 to -4.2 kg/m², respectively, compared to -3.0 cm and -0.84 kg/m² in the placebo group. In addition to weight loss, significant improvements in other cardiovascular and metabolic indicators, such as glucose metabolism, blood pressure, and blood lipids, were also observed with HDM1005.
In the aforementioned Phase II study, HDM1005 demonstrated good safety and tolerability. The majority of adverse events were mild to moderate in severity. No adverse events led to permanent discontinuation or early withdrawal from the trial, and no treatment-related serious adverse events occurred. Target-related adverse events such as acute cholecystitis, acute pancreatitis, and acute kidney injury did not occur. The most common adverse reactions were decreased appetite and gastrointestinal adverse reactions (diarrhea, nausea, vomiting, and abdominal distension). The incidence rates of nausea in the HDM1005 0.5 mg to 4.0 mg dose groups and the placebo group were 4.0%, 8.3%, 2.1%, 18.8%, and 2.0%, respectively. The incidence rates of vomiting were 6.0%, 6.3%, 0%, 12.5%, and 4.1%, respectively.