CAR-T Cell Immunotherapy: Technical Advantages, Risks, and Key Application Scenarios
Produced independently by Probe Capital. Discussions are welcome—please add WeChat ID: S-caffeine
I. Principles of CAR-T Technology
(1) Adoptive Cell Therapy
Adoptive Cell Therapy Is a Major Research Direction in Cellular Immunotherapy. Adoptive Cell Transfer (ACT) is a form of passive immunotherapy for cancer, which involves isolating autologous tumor-infiltrating lymphocytes (TILs) or peripheral blood lymphocytes from cancer patients, followed by in vitro selection, expansion, and activation, and then reinfusing them into the patient’s body.
In malignant melanoma and renal cell carcinoma, tumor-infiltrating lymphocytes (TILs) are a commonly used form of adoptive cell therapy (ACT). TILs are lymphocytes that infiltrate tumors; they are isolated, expanded ex vivo in the presence of factors such as interleukin-2 (IL-2), and then reinfused into the patient. However, TIL therapy is not applicable in most cases: some patients lack tumor specimens, or have few TILs within the primary tumor and metastatic lesions; obtaining fresh tumor tissue and isolating and expanding TILs is technically challenging; reinfused TILs often exhibit impaired function and fail to effectively recognize tumor cells in vivo; and the potent immunosuppressive microenvironment within tumors diminishes the cytotoxic capacity of the reinfused cells. These issues limit the widespread application of TIL therapy, which has demonstrated efficacy only in a limited number of cancer types, such as malignant melanoma and renal cell carcinoma, while showing suboptimal effectiveness in most other tumors.
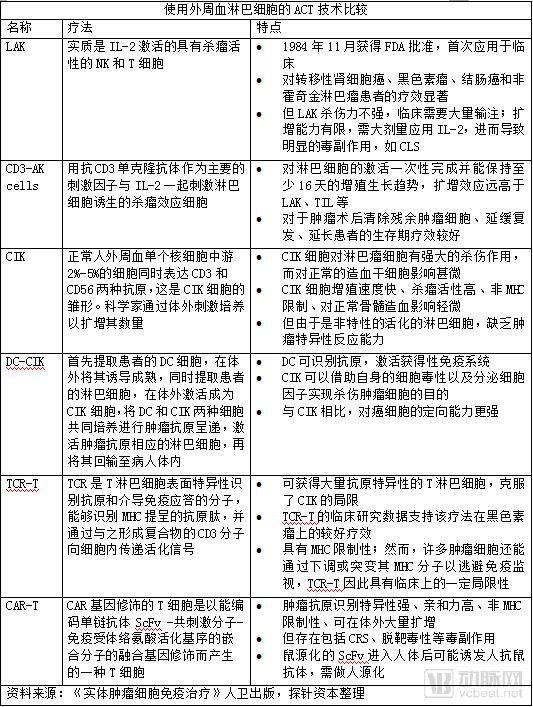
In this context, adoptive cell therapy (ACT) requires the use of peripheral blood lymphocytes. Current ACT approaches utilizing peripheral blood lymphocytes include lymphokine-activated killer (LAK) cells, cytokine-induced killer (CIK) cells, dendritic cell–cytokine-induced killer (DC-CIK) cells, and anti-CD3 antibody-induced activated killer (CD3-AK) cells. However, CIK cells and similar populations are non-specifically activated lymphocytes that lack tumor-specific reactivity.
In this context, scientists have begun to use genetic modification to introduce T cell receptor (TCR) or chimeric antigen receptor (CAR) genes that recognize tumor antigens into lymphocytes, thereby generating TCR-engineered T cells (TCR-T) or CAR-T cells with the ability to specifically target and recognize tumor antigens.
(II) The Treatment Process of CAR-T
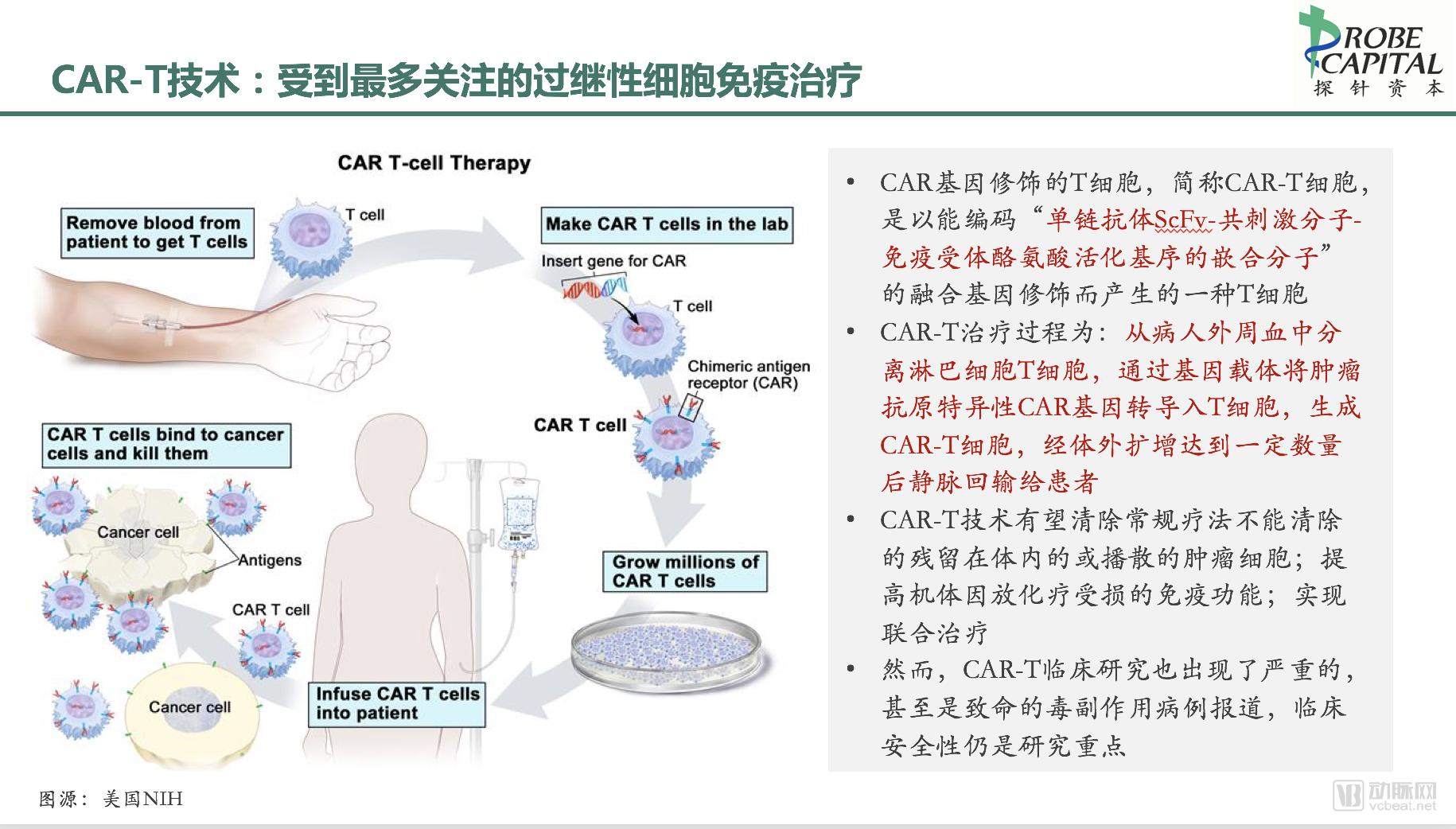
In brief, the CAR-T therapy process involves isolating T lymphocytes from the patient’s peripheral blood, transducing them with a tumor antigen-specific chimeric antigen receptor (CAR) gene via a genetic vector to generate CAR-T cells, expanding these cells ex vivo to a sufficient quantity, and then reinfusing them intravenously into the patient. Due to the functional properties conferred by the transduced CAR gene, the reinfused CAR-T cells can specifically bind to cancer cells, thereby achieving targeted therapeutic effects.
(3) Structure of CAR-T
1.3.1 Overview of CAR Structure
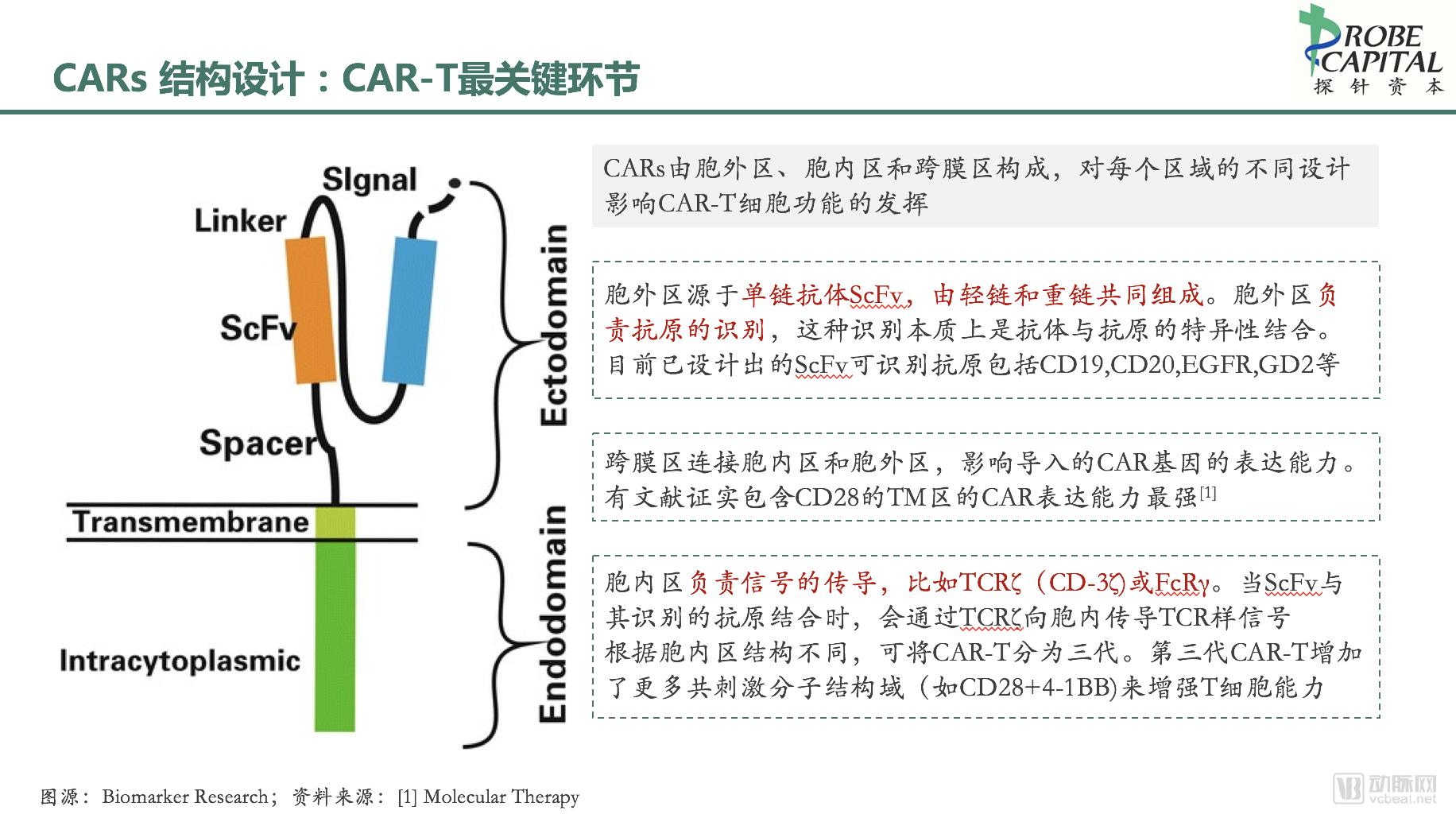
CARs are recombinant receptors that target surface molecules on the intended cells. Their structure primarily consists of three parts: an extracellular antigen-binding domain, a transmembrane region, and an intracellular signaling domain. Different designs for each region influence the functional efficacy of CAR-T cells.
1.3.2 Extracellular Antigen-Binding Domain
The extracellular domain is derived from a single-chain variable fragment (scFv), composed of light and heavy chains, and is responsible for antigen recognition. This recognition essentially involves the specific binding between an antibody and an antigen, independent of MHC presentation, thereby effectively circumventing the immune escape mechanism of downregulated MHC expression in tumor cells. CARs can recognize not only peptide antigens but also carbohydrate and glycolipid antigens, enabling broader-spectrum killing of tumor cells.
Currently designed scFvs can recognize antigens including CD19, CD20, EGFR, Her2/neu, GD2, PSMA, and ROR1.
1.3.3 Transmembrane Domain
The transmembrane domain connects the intracellular and extracellular domains, typically consisting of dimeric membrane proteins, and anchors the CAR structure to the T cell membrane.
Variations in transmembrane domain design influence the expression levels of introduced CAR genes. Literature has confirmed that CARs incorporating the CD28 transmembrane domain exhibit the strongest expression, followed by those containing OX40, while CARs with the CD3ζ transmembrane domain show the weakest expression. Currently, transmembrane domains commonly designed for CARs primarily include H2-Kb, CD4, CD7, CD8, and CD28.
1.3.4 Intracellular Signaling Domain
The intracellular signaling domain employs immune-receptor tyrosine-based activation motifs (ITAMs), typically derived from TCRζ (CD3ζ) or FcRγ. Upon binding of the extracellular scFv to its target antigen, TCR-like signals are transduced into the cell. Based on differences in intracellular domain structure, CAR-T cells are classified into four generations.
1.3.5 Structural Evolution of CARs
Based on Differences in Intracellular Domain Structures, CAR Architectures Have Evolved from First- to Fourth-Generation Designs.
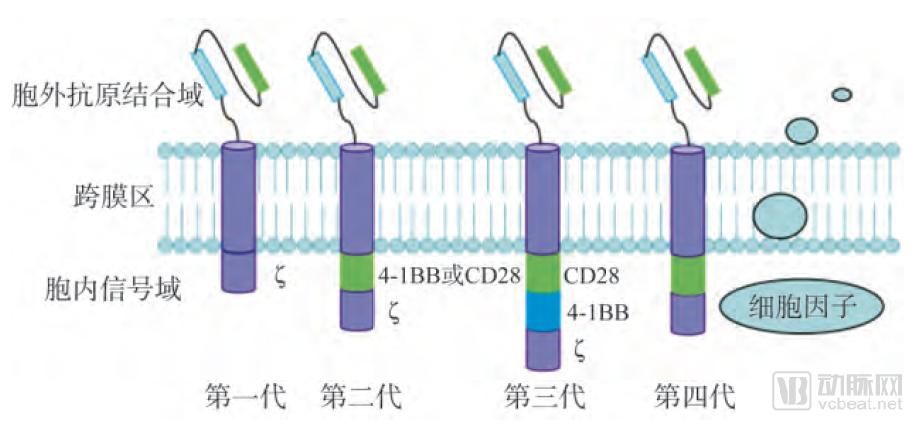
Image source: CNKI
First-generation CARs contain only a single intracellular signaling component, primarily CD3ζ or FcRγ. Upon recognition and activation by specific antigens, first-generation CARs provide an activation signal to T cells and transmit this signal through the intracellular domain, leading to cellular activation. This manifests as CAR-dependent T cell activation and cytotoxicity, characterized by the secretion of perforin, granzymes, and cytokines, which work synergistically to kill tumor cells. Early experiments demonstrated the feasibility of CAR-T therapy, marking the first attempt to activate T cells independently of human leukocyte antigen (HLA). However, first-generation CARs induce only transient T cell activation and secrete low levels of cytokines; they fail to provide sustained signals for T cell expansion or durable in vivo antitumor effects.
To address this issue, costimulatory molecule (CM) signaling domains, such as CD28 and CD137 (4-1BB), were introduced starting with second-generation CARs. Compared with first-generation CARs, second-generation CARs incorporate one activation domain and one costimulatory domain, thereby enhancing T-cell proliferation and cytokine secretion while maintaining consistent antigen specificity.
Third-generation CARs represent an upgrade over previous versions. Their intracellular domain consists of an activation domain and multiple co-stimulatory regions, comprising three intracellular signaling domains: two tandem co-stimulatory domains (CD28, 4-1BB, or OX40) and one CD3ζ domain. The addition of these domains not only enhances the ability of CAR-T cells to specifically recognize and bind to tumor antigens but also significantly amplifies cellular signals transmitted from the extracellular region, triggering a cascade amplification of downstream cytotoxic effects, thereby resulting in superior anti-tumor efficacy.
Fourth-generation CARs are a newly emerged type of CAR, also known as TRUCKs (T cells redirected for universal cytokine killing). Their structure differs from that of the first three generations by incorporating pro-inflammatory cytokines (such as IL-12) and co-stimulatory ligands (4-1BBL and CD40L). In the immunosuppressive tumor microenvironment, they can release pro-inflammatory factors to recruit and activate additional immune cells, thereby eliciting a broader anti-tumor immune response. Furthermore, the advent of fourth-generation CARs spares patients from the adverse effects associated with pre-infusion conditioning regimens (such as total body irradiation or high-dose chemotherapy), reduces the total number of infused cells, and expands the clinical application scope of CAR-T cell therapy.
(4) Antibody Structure of CAR-T
1.4.1 Traditional Antibodies
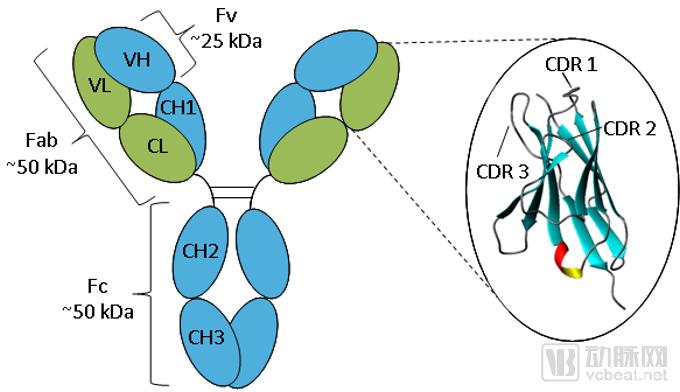
Image source:Absolute Antibody
Traditional anti-The antibody is a tetrameric polypeptide with a molecular weight of 150 kDa, composed of two pairs of identical heavy chains (50 kDa) and light chains (25 kDa). These chains are linked by interchain disulfide bonds, forming a Y-shaped or T-shaped structure. The light chain consists of a variable region at the N-terminus (VL) and a constant region at the C-terminus (CL); the heavy chain comprises a variable region at the N-terminus (VH) and three to four constant regions (CH1, CH2, CH3, and potentially CH4).
The diversity of the variable regions determines the diversity and specificity of antibodies, enabling them to bind to specific antigens. Comparison of the amino acid sequences of VL and VH among antibodies with different specificities reveals that variations are concentrated in only a few regions (15%–20%), known as hypervariable regions. These hypervariable regions constitute the antigen-binding sites of antibodies and are structurally complementary to antigenic determinants; hence, they are also referred to as complementarity-determining regions (CDRs).
The extracellular antigen-binding domain is derived from the antigen-binding motif of antibodies, which can link the VH and VL regions on the receptor to form single-chain variable fragments (scFvs), thereby specifically recognizing tumor antigens.
1.4.2 Nanobodies
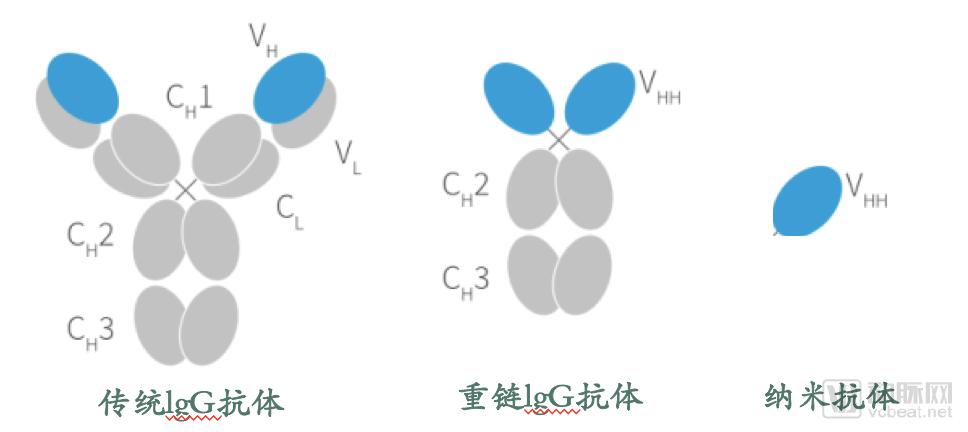
Camels and shark-like animals possess heavy-chain antibodies that naturally lack light chains, composed of two homologous heavy chain polypeptides with a relative molecular mass of 90 kDa. Cloning their variable region (VHH) yields single-domain antibodies consisting solely of a single heavy-chain variable domain. These represent the smallest currently available functional, stable, antigen-binding units, also known as nanobodies, with a relative molecular mass of only 15 kDa, approximately one-tenth that of conventional antibodies.
The VHH domain of nanobodies is structurally very similar to the variable heavy (VH) domain of human antibodies, with a gene homology of 90%. The surface of the nanobody VHH differs from that of human VH by only approximately 10 amino acids. Among these, four specific amino acid substitutions occur in framework region 2 (FR2). In conventional antibodies, the residues V37, G44, L45, and W47 in FR2 are highly conserved hydrophobic residues throughout evolution. In contrast, in VHH domains, these residues are mutated to hydrophilic amino acids F37, E44, R45, and G47, thereby enhancing the solubility of VHH.
Cysteine (Cys) residues are present in the CDR1 and framework region 2 (FR2) of VHHs, where they can form disulfide bridges with Cys residues in the CDR3 region, thereby enhancing the stability and structural rigidity of the variable domain. The substitution of specific hydrophilic amino acid residues in the FR2 region of VHHs, combined with the presence of disulfide bonds in the CDR3 region, confers significantly superior stability and hydrophilicity compared to conventional antibodies.
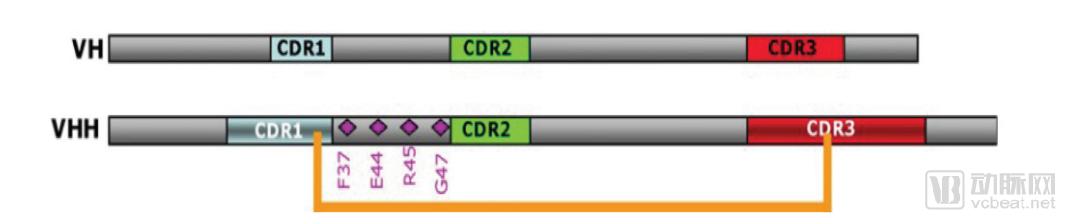
Schematic Diagrams of VH and VHH Structures
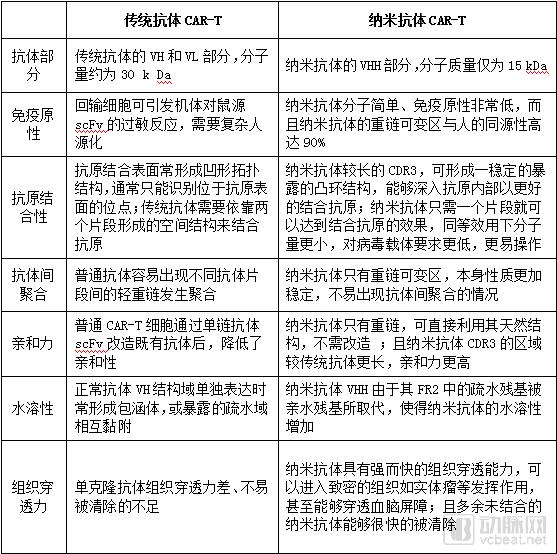
Compared with traditional CAR-T, CAR-T equipped with nanobodies has more advantages in performance.
(V) Gene Modification Methods for CAR-T
Stable transfection is a prerequisite for the stable expression of chimeric antigen receptors (CARs) in effector T cells. Currently, the primary methods for gene transduction in CAR-T therapy include retroviral transduction, lentiviral transduction, and mRNA transfection.
The Retroviridae family comprises seven members, among which gammaretroviruses have become commonly used vectors for clinical gene transfer in the treatment of malignant tumors. However, in recent years, retroviral transduction has been gradually replaced by lentiviral transduction in clinical trials due to concerns such as the risk of insertional oncogenesis, the inability to infect non-dividing cells, and low viral titers.
Lentiviral vectors contain the cis-acting central polypurine tract (cPPT) element, enabling transduction that is independent of cell division. This allows for efficient transduction across a broader range of cell types, including non-dividing (quiescent) cells. Furthermore, lentiviral vectors offer safer integration sites, higher transgene cargo capacity, and reduced genotoxicity. In clinical trials of CD19-targeted CAR-T therapy, CAR-T cells transduced with lentiviral vectors have demonstrated more potent anti-tumor efficacy.
mRNA transfection is employed when transiently existing CAR-T cells are required. As a rapid and efficient transfection method, mRNA transfection enables T cells to express the chimeric antigen receptor transiently within one week without stable, long-term persistence. Consequently, large quantities of T cells often need to be infused to ensure therapeutic efficacy.
(VI) Advantages of CAR-T Technology
Overall, current CAR-T therapies hold advantages over TIL and TCR therapies in terms of principle, structure, and manufacturing process.
1. CAR-T cells are not restricted by MHC; they rely on the specific recognition between antigens and antibodies, enabling more effective killing of tumor cells with antigen specificity. In contrast, both TILs and TCRs can only recognize antigens presented by MHC molecules. Consequently, tumor cells may evade immune surveillance by downregulating or mutating their MHC molecules, leading to certain clinical limitations.
II. The dosage of CAR-T cell therapy is two to three orders of magnitude lower than that of TIL and TCR-T therapies. Due to its well-defined targets and high specificity in recognizing tumor surface antigens, as well as its ability to overcome MHC restriction, CAR-T therapy requires a significantly lower number of cells per infusion (10 million to 100 million) to achieve comparable therapeutic efficacy, compared with TCR-T (1 billion to 10 billion) and TIL (10 billion to 150 billion).
III. CAR-T Cell Therapy Requires Less Time. Since CAR-T therapy requires fewer cells to achieve equivalent therapeutic efficacy, the time needed for T-cell culture is the shortest. The ex vivo cultivation period has been reduced to two weeks, significantly saving time costs.
IV. CARs can recognize not only peptide antigens but also carbohydrate and glycolipid antigens, thereby expanding the range of tumor antigen targets. In addition to being unrestricted by MHC, CAR-T therapy is not limited by protein antigens on tumor cells; CAR-T cells can leverage non-protein glycolipid antigens on tumor cells, enabling multidimensional antigen recognition.
V. CAR-T possesses a certain degree of broad-spectrum reproducibility. Since certain targets, such as EGFR, are expressed in various types of tumor cells, the CAR gene constructed against such antigens can be widely utilized once developed.
VI. CAR-T cells possess immunological memory function and can survive in the body for a long term. This holds significant clinical importance for preventing tumor recurrence.
(7) Risks and Solutions of CAR-T Technology
1.7.1 Off-Target Toxicity
Off-target toxicity is a common issue in all adoptive cell immunotherapies, and its concept should be distinguished from on-target toxicity.
On-target toxicity refers to the toxicity of genetically modified T cells against normal tissues that also express their targets. Off-target effects refer to the toxicity of genetically modified T cells against normal tissues or organs that do not express these target molecules.
Identifying suitable immunogenic targets on tumor cells to enable CAR-T cells to selectively attack tumors without damaging normal tissues is key to the development of CAR-T therapy. One approach involves designing CAR structures that target multiple antigens simultaneously; by recognizing specific antigen combinations, this strategy enhances killing specificity. For instance, the Wilkie laboratory employed CAR-T cells co-targeting ErbB2 and MUC1 for the treatment of breast cancer.
1.7.2 Cytokine Storm (Cytokine Release Syndrome, CRS)
When CAR-T cells are infused back into the body, a large amount of cytokines, including IL-6, TNFα, and IFNγ, are released during the cell therapy process. These inflammatory mediators trigger an acute inflammatory response that induces epithelial and tissue damage, leading to various immune-inflammatory reactions in the body such as microvascular leakage, a condition known as cytokine release syndrome (CRS). CRS occurs in more than half of CAR-T trial participants and is one of the most common adverse events.
CRS can occur 2–3 weeks after cell infusion. Common symptoms include fever, chills, fatigue, hypotension, nausea, headache, tachycardia, dyspnea, and cardiac, hepatic, and renal dysfunction.
The occurrence of cytokine release syndrome (CRS) is associated with the structure of the chimeric antigen receptor (CAR), tumor burden and type, as well as patient genetic polymorphisms. The risk can be reduced by designing safer CARs and strictly limiting the number of cells infused per administration. Alternatively, a safety switch control gene, such as inducible caspase 9, can be introduced; when induced by a specific soluble factor, this gene triggers apoptosis of CAR-T cells, thereby helping to mitigate cellular toxicity. Furthermore, once CRS occurs, it can be treated and controlled using medications such as tocilizumab.
1.7.3 Neurotoxicity
Another common adverse reaction is neurotoxicity. Following the successful use of tocilizumab to treat cytokine release syndrome (CRS), neurotoxicity has become the primary life-threatening event during CAR-T therapy, and its underlying mechanisms remain to be elucidated.
Multiple clinical trials have reported a variety of neurological abnormalities, including cerebral infarction, delirium, impaired consciousness, convulsions, neural paralysis, visual field defects, ataxia, and speech disorders.
U.S.-based Juno Therapeutics was forced to announce the termination of clinical trials for its “Rocket Project” after five unexpected deaths due to cerebral edema occurred during the trials. Juno’s analysis suggested that the neurotoxicity might have been caused by the pre-conditioning chemotherapy drug fludarabine or the CD28 co-stimulatory domain in the CAR-T cell product structure.
1.7.4 Other Toxic Side Effects
CAR-T therapy is also associated with certain other toxic side effects, such as coagulation disorders and B-cell dysplasia.
Coagulation disorders mainly manifest as scattered ecchymoses and petechiae, thrombosis, and abnormal laboratory indicators, such as thrombocytopenia, elevated D-dimer, decreased fibrinogen, elevated fibrin degradation products, and prolonged activated partial thromboplastin time.
B-cell dysplasia primarily occurs because the targets for tumor cells (such as CD19) are also expressed on the surface of normal B cells, leading CAR-T cells to eliminate both tumor and normal B cells. Furthermore, since CAR-T cells can persist and remain active in the body for an extended period, patients may experience a long-term deficiency of normal B cells, resulting in hypogammaglobulinemia and increased susceptibility to infections. Therefore, patients require appropriate immunoglobulin supplementation to enhance immunity as a preventive measure.
When CAR-T therapy is applied to the treatment of solid tumors, it may also lead to issues such as impaired CAR-T cell homing and an immunosuppressive tumor microenvironment.