Over 10 New Drugs/New Indications Approved in China; Domestic Innovations Secure Over $2 Billion in Global Licensing Deals

Johnson & Johnson
Medical Device R&D and Manufacturer

AstraZeneca
Pharmaceutical Technology Research and Development Provider

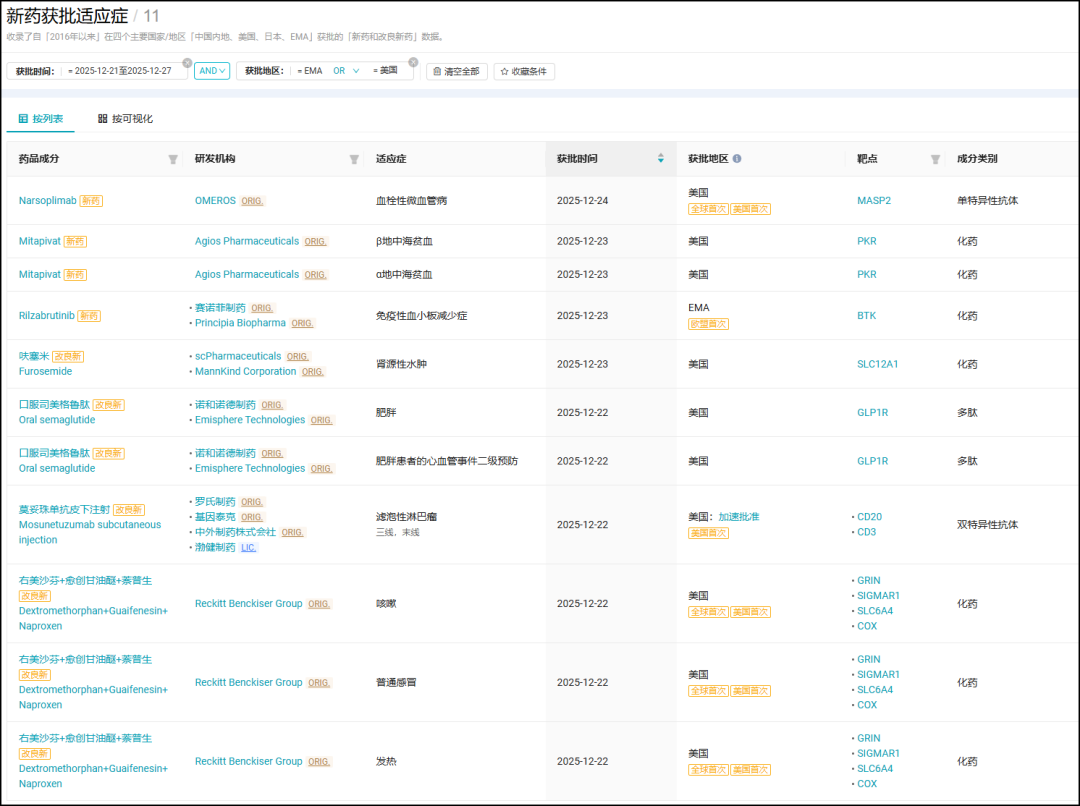
According to the "Global New Drug" module of the Insight database, this week (December 21 - December 27), a total of 117 innovative drugs (including improved new drugs) worldwide have advanced to a new stage of development, of which 3 were approved for the first time, 4 were submitted for marketing authorization for the first time, and 8 registered for Phase III clinical trials for the first time., 33 new Phase I clinical trial registrations.
The following text, Insight will introduce the progress of some key projects at home and abroad this week.
*Data Description: The data collection time for this weekly report is 2025-12-28 11:00. Due to the continuous high-speed updates of the Insight database, there may be differences in search results at different times. Please refer to the latest query for accuracy.

1February 22, RocheAnnounced that the FDA has approved CD20/CD3 Bispecific Antibody Subcutaneous Injection FormulationLunsumio VELO™(Motelizumab Subcutaneous InjectionLaunched for the treatment of relapsed or refractory patients who have received at least two prior systemic therapies.(R/R)Follicular Lymphoma(FL)。
Based onResults of the Phase I/II GO29781 Study,Motelizumab Subcutaneous Injection Receives FDA ApprovalAccelerated Approval,Full approval may depend on the verification and confirmation of efficacy in confirmatory trials.In November, this indication received conditional approval from the EU.

Source: Roche Official Website
Compared with intravenous infusion which takes 2-4 hours,Moxetumomab subcutaneous injection can be administered in approximately 1 minute., greatly shortening the treatment time. Compared with intravenous injection,MotelizumabThe same,Motelizumab Subcutaneous InjectionIt can also be administered in an outpatient setting, with a fixed course of treatment, and the shortest duration can be six months.
The FDA approval is based onPrimary analysis results of the GO29781 study. This study evaluatedEfficacy of Subcutaneous Moxetumomab in Patients with Third-Line and Above Follicular Lymphoma. The results showed that patients receivingSubcutaneous Injection Formulation of Motolimab for TreatmentPatient Objective Response Rate(ORR)And Complete Remission Rate(CR)At 75% and 59%, respectively, with a median duration of response of 22.4 months.
The most common adverse reactions include injection site reactions, fatigue, rash, and cytokine release syndrome.COVID-19 infection, musculoskeletal pain, and diarrhea.
Local time on December 22Viridian Therapeutics Announces that the U.S. FDA Has Accepted Its Application for the Treatment ofThyroid Eye Disease(TED)VEligrotug Biologics License Application(BLA). The application has been granted priority review status,PDUFA Date: June 30, 2026。

The BLA for Veligrotug is supported by positive data from the two largest Phase III clinical trials for TED conducted to date. In the THRIVE and THRIVE-2 trials, which were conducted in patients with active and chronic TED respectively, Veligrotug met all primary endpoints of each study as well as all secondary endpoints, demonstratingQuick OnsetClinical benefits.
Veligrotug Shows Statistically Significant Results for the First Time in Phase III Clinical Trials for Chronic TEDRelief and Resolution of DiplopiaIf approved, Veligrotug will offer patients an extremely attractive treatment option.A course of five infusions allows patients to complete treatment within 12 weeks.Veligrotug was generally well-tolerated in the Phase III clinical trial.
Veligrotug(VRDN-001)YesAn Intravenous Anti-Insulin-Like Growth Factor-1 Receptor(IGF-1R)Antibody, expected to become a promising treatment for patients with active and chronic thyroid eye disease.Preferred Intravenous Treatment Regimen。
October 2020,Zenith(Zenas BioPharma)Granted an exclusive license from Viridian Therapeutics to develop, manufacture, and commercialize in Greater China.Veligrotug And other drugs targeting IGF-1R for the treatment of non-oncology indications. In 2024,ZenathWillVeligrotug The Greater China rights have been licensed to Zai Lab.
On December 22, AstraZeneca announcedFinishedceralAsertib CombinationDurvalumab for the Treatment of Locally Advanced or Metastatic Non-Small Cell Lung Cancer(NSCLC)Phase IIILATIFY Latest Developments. The results showed that the combination regimen failed to achieve overall survival(OS)Primary endpoint.

Source: AstraZeneca Official Website
In terms of safety,ceralAsertib CombinationDurvalumabOverall, the treatment was well-tolerated, and the safety profile was consistent with the known safety of each drug individually, with no new safety issues identified. These data will be presented at an upcoming medical conference.
LATIFY is aRandomized, Open-label, Multicenter, Global Phase III Clinical Trial Aimed at Evaluating Ceralasertib UnitedEfficacy of Durvalumab in Locally Advanced or Metastatic NSCLC Patients Without Actionable Genomic Alterations (AGA) and Previously Treated with Anticancer TherapyProgression of the disease after PD-(L)1 treatment and platinum-based chemotherapy.
The study included a total of594 subjects from more than 20 countries, randomly assigned to receiveCeralasertib 240mg Oral Tablets(Twice daily for 7 consecutive days)UnitedDurvalumab1500mg Fixed Dose(Day 8 of taking), repeated every 4 weeks; or receive docetaxel treatment, repeated every 3 weeks, until disease progression, intolerable toxicity occurs, the patient withdraws informed consent, or the criteria for terminating the trial are met.
The primary endpoint of the study isOS, secondary endpoints include progression-free survival, objective response rate, duration of response, time to response, disease control rate, and patient-reported outcomes.
CeralAsertib is aOral Johnson & JohnsonEfficacious Selective ATR Inhibitor,Acting on the tumor microenvironment, when used in combination with immunotherapy, it can shift from an immunosuppressive state to an immune-activated state.
DXY Insight database shows that currently under researchATR inhibition has 40 candidates under research, ceralAsertib is the fastest progressing globally. In China, Hengrui, Takeda Pharmaceutical,Zhikang Hongyi, Yingpai Pharmaceuticals, and Fosun Pharma are all in development.

Pharmaceutical Transactions
According to the Insight database, a total of 32 transaction events occurred this week (December 21 - December 27).
On December 23, Tongyi Pharmaceuticals announced today that it has entered into an agreement with MultiValent Biotherapies, Inc. for the treatmentProstate CancerThe polypeptide conjugate drug CBP-1018 has reached an exclusive licensing agreement.

Screenshot source: Official WeChat account of the company
According to the terms of the agreement, MultiValent will obtain an exclusive license to develop and commercialize CBP-1018 in markets outside of Greater China. In return, Tongyi Medicine will receive$20 millionThe down payment and MultiValent company20% of the equity。
In addition, CytRx Pharmaceuticals also has a future cumulativeUp to approximately USD 2 billionDevelopment, regulatory and commercial milestone payments, as well as tiered sales royalties.
CBP-1018 is a Bi-XDC proprietary product developed by Tongyi Pharmaceutical.(Bispecific Dual-Ligand Conjugate Drug)The second innovative drug developed by the platform. CBP-1018 is a bispecific peptide drug conjugated with Auristatin-E, targeting two proteins highly expressed in prostate cancer cells: PSMA and FRα.
In a Phase I/II clinical trial involving more than 110 patients with metastatic castration-resistant prostate cancer, intravenous administration of CBP-1018 demonstrated encouraging preliminary safety and efficacy. Across all dose groups, CBP-1018 achievedMedian Progression-Free Survival of 8.5 Months(mPFS)`, this data`Comparable to the published mPFS data of the currently only approved PSMA-targeted radioligand therapy。
December 22, Quanxin BioAnnounced that it has reached a licensing agreement with LE2025, an affiliate of Windward Bio, for QX027N.Total amount up to 700 million US dollars。
This is Quan.The third overseas deal reached by Xinyi Biotech this year. Previously, it granted Caldera the global rights to QX030N.The global rights of QX031N have been granted to Roche.

Source: Quanxin Biotech Announcement
According to the agreement, LE2025 has obtained globally(Excluding mainland China, Taiwan, Hong Kong Special Administrative Region, and Macao Special Administrative Region)The exclusive rights to develop and commercialize QX027N in China. In return, Quanxin Biotech will be entitled to receiveUp to $700 Million in Payments, including the upfront payment, equity in Windward Bio, development and commercial milestone payments. In addition, the company is entitled to receive royalties based on the net sales of QX027N in the licensed territory.Tiered Royalty。
QX027N is independently developed by Quanxin Biotechnology.Long-acting anti-TSLP x IL-13 bispecific antibody, November 2025 has received the clinical trial implied permission from CDE(Application No.: CXSL2500757/8), intended for the treatment of asthma and atopic dermatitis, has now completed the enrollment of the first subject in Phase I clinical trials in China.
On December 21, JACOBIO announced that it has reached an agreement on its self-developedPan-KRAS(Pan-KRAS)Inhibitor JAB-23E73Reached a cooperation agreement with AstraZeneca,Total amount up to 2.015 billion US dollars。
AstraZeneca will obtain the exclusive rights for the development and commercialization of this product in markets outside of China. In the Chinese market, Jacobio will co-develop and co-commercialize the product with AstraZeneca.

Source: JACOBIO Announcement
According to the terms of the agreement, Jacobio will receive$100 million upfront payment, and is eligible for additionalUp to $1.915 billion in development and commercialization milestone payments, as well as tiered royalties on net sales achieved in markets outside of China. AstraZeneca will be responsible for all clinical development, regulatory submissions, and commercialization activities for JAB-23E73 in markets outside of China.
JAB-23E73 is an innovative pan-KRAS inhibitor developed by JACOBIO based on its induced allosteric platform, designed to target multiple KRAS mutant subtypes. KRAS is the most frequently mutated oncogene in humans, accounting for approximately 23% of all patients. JAB-23E73 is currently undergoing Phase I clinical trials in China and the United States, with early signals of anti-tumor activity already observed.


Ricalto®, Rebyline®, and Mebirel® are independently developed based on Luye Pharma's long-acting and sustained-release technology platform. The active ingredient of Ricalto® is risperidone, and this product is...China's FirstSecond-generation antipsychotic long-acting injectable developed through independent innovation. The active ingredient in both Ribilai® and Mebilai® is paliperidone palmitate. Among them, Ribilai® isThe World's FirstThe only paliperidone extended-release injectable monthly formulation that achieves the "only one injection needed in the first month" dosing regimen.
According to the agreement, Enhua Pharmaceuticals will be fully responsible for the exclusive distribution and commercialization of these three long-acting injectable products in mainland China. Luye Pharma will continue to hold the asset rights, registration certificates, all intellectual property rights, and other rights of these three products, as well as being responsible for their production and supply. The cooperation period will start from the effective date of the agreement.Until December 31, 2035。
In addition, Enhua Pharmaceutical needs to make a one-time payment$20 millionNon-refundable authorization consideration, and a commitment to complete the cooperative products between 2026 and 2035.Total not less than 2.7 billion yuanSales in RMB, including tax.

On December 25, Johnson & Johnson announced the innovative therapeutic drug Rikojie® - Amivantamab Injection(Subcutaneous Injection)Officially received NMPA approval in China for the treatment of patients with epidermal growth factor receptor(EGFR)Advanced Non-Small Cell Lung Cancer with Mutations(NSCLC)Patient.
The indications for which Rapihex® has been approved for marketing in China include:
1) Lanzetinib in combination is indicated for the first-line treatment of adult patients with locally advanced or metastatic NSCLC harboring EGFR exon 19 deletions or exon 21 L858R substitution mutations.
2) In combination with carboplatin and pemetrexed, it is indicated for the treatment of patients with EGFR exon 19 deletions or exon 21 L858R substitution mutations who have progressed on EGFR tyrosine kinase inhibitors. (TKI) Adult patients with locally advanced or metastatic non-squamous NSCLC whose disease has progressed during or after treatment;
3) In combination with carboplatin and pemetrexed, it is indicated for the first-line treatment of adult patients with locally advanced or metastatic NSCLC whose tumors have EGFR exon 20 insertion mutations as detected by a validated test.
Amivantamab is a fully human EGFR×MET bispecific antibody. The subcutaneous injection formulation of Amivantamab contains recombinant human hyaluronidase.(rHuPH20), using Halozyme's ENHANZE® drug delivery technology,Administration time is only about 5 minutes.,Compared with intravenous injections, the incidence of infusion-related adverse reactions was reduced by 80%.。
This approval was based on the positive results of the PALOMA-3 Phase III study and the PALOMA-2 Phase II study.
PALOMA-3 is a randomized, open-label Phase III study. It evaluates the non-inferiority in pharmacokinetics of subcutaneous amivantamab combined with lazertinib versus intravenous amivantamab combined with lazertinib in patients with EGFR-mutated locally advanced or metastatic NSCLC whose disease progressed during or after treatment with osimertinib and platinum-based chemotherapy. The study results showed that the subcutaneous formulation of amivantamab achieved blood drug concentration levels meeting the two co-primary pharmacokinetic endpoints, demonstrating its non-inferiority compared to the intravenous formulation.
PALOMA-2 is an open-label, parallel-cohort study designed to evaluate the efficacy, safety, and pharmacokinetic profile of subcutaneous Amivantamab in patients with locally advanced or metastatic NSCLC carrying EGFR mutations. The study results show that the subcutaneous formulation of Amivantamab has comparable efficacy to the previously approved intravenous formulation, while offering advantages in tolerability and ease of administration.
Patients receiving amivantamab subcutaneous injection combined with lazertinib had an incidence of infusion-related adverse reactions only one-fifth that of the intravenous injection group.(13% vs 66%)Subcutaneous injection formulation group: Incidence of venous thromboembolism in patients with and without prophylactic anticoagulation therapy(VTE)The incidence rates were 7% and 17%, respectively. Apart from the incidence rates of IRR and VTE, the overall safety profile of the subcutaneous formulation of amivantamab was consistent with that of the intravenous formulation.

According to the Insight database, Pasireotide was first approved for marketing in the United States on August 28, 2020, becomingThe World's FirstFor adult growth hormone deficiency(AGHD)Long-acting formulation;It was then approved for expanded use in pediatric patients in the United States in April 2023.
Novo Nordisk has registered two Phase III clinical trials for Pasireotide in China, REAL6(NCT04970654)AndREAL8(NCT05330325), respectively targeting different patient populations for research, and have currently announced clinical results.
REAL6 StudyIncluded 110 cases who had not received growth hormone(GH)Chinese prepubertal GHD patients were randomly assigned in a 2:1 ratio to receive weekly subcutaneous injections of Pasitropin.(0.16 mg/kg;n=74)Oral GH Daily Formulation Group(0.034 mg/kg;n=36), the treatment duration was 52 weeks.
The 52-week results showed that, among the 103 Chinese GHD children who completed the treatment, the efficacy and safety of Pasitropin were comparable to those of daily GH formulations. The children in the Pasitropin treatment group...The estimated average growth rate reaches 11.0 cm/year., GH daily formulation group was 10.4 cm/year.

According to the terms of the agreement, Abbisko Therapeutics will receive$70 millionA one-time, non-refundable upfront payment. If Merck exercises the global commercialization option, Abbisko will also receive an additional exercise fee; including R&D milestone payments and sales milestone payments, the total potential payments may reach up to$605.5 millionIn addition, Merck will pay Abbisko Therapeutics a double-digit percentage of sales royalties.
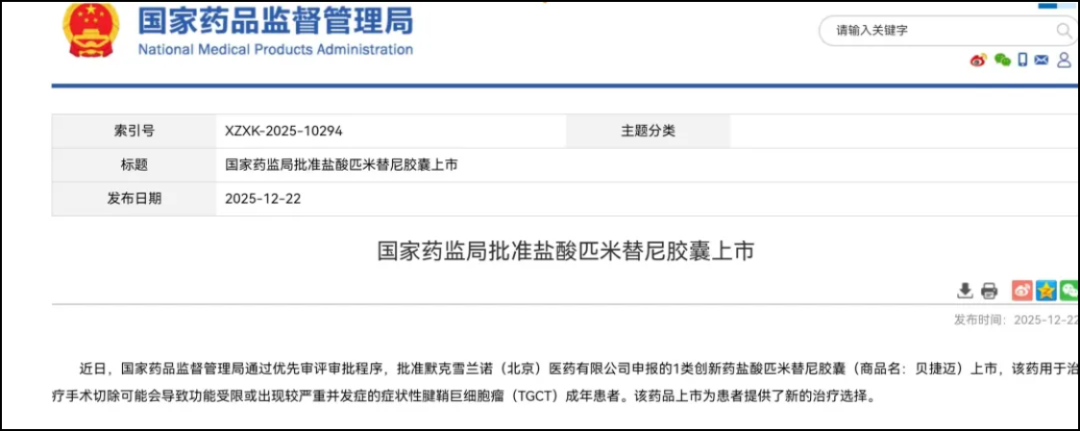
On April 1, 2025, Abbisko Therapeutics announced that Merck had, according to the previously signed licensing agreement,Exercise the global commercialization option for Pimitinib, with an exercise fee of $85 million。
In November 2024, Abbisko Therapeutics announced that the Phase III MANEUVER study of Pimicotinib for the treatment of TGCT met its primary endpoint.Objective Response Rate at Week 25(ORR)For 54.0%, while the placebo was 3.2%。
In2025 International Society for Connective Tissue Oncology(CTOS)At the annual meeting, Abbisko Therapeutics announcedMANEUVER StudyLong-term follow-up data.
The latest analysis data shows that at a median follow-up of 14.3 months:
According to the Blinded Independent Review Committee(BICR)Assessed by RECIST v1.1 criteria, received from the start of the studyPimitespibTreating PatientsORR Significantly Increased from 54% at Week 25 to 76.2%;
Endpoint Metric Specifically Designed for TGCT —— BICR Tumor Volume Score(TVS)AssessmentORR Increased from 61.9% at Week 25 to 74.6%;
At the time of data cutoff, the median duration of response had not yet been reached, and long-term follow-up revealed that,93.7%AcceptPimitespibPatients were treated and assessed by BICR using RECIST v1.1 criteria.The tumor volume was reduced.。
Moreover, in a long-term follow-up of up to 73 weeks, pimitinib showed significant effects on key patient-reported outcomes impacting the quality of life for TGCT patients.(Including range of motion, pain, stiffness, and physical function)In terms of clinical significance, improvements were demonstrated.
Romosozumab(Romosozumab)YesDeveloped jointly by Amgen and UCBAn innovative monoclonal antibody drug,Previously, the drug has been approved for marketing in Japan, the United States, and the European Union for the treatment ofPostmenopausalOsteoporosis。
FDA on RomosozumabThe approval was based on the results of two Phase III studies. In the placebo-controlled FRAME study, compared with placebo, patients usingRomosozumabAfter 12 months of treatmentSignificant Reduction in New Spinal Fractures. Compared with women transitioning from placebo to denosumab, during the first year of receivingRomosozumabWomen Treated and Transitioned to DenosumabStill significantly reduces the risk of fractures in the second year。
Moreover, compared with patients who received placebo treatment for one year,RomosozumabSignificantly increased bone mineral density in the lumbar spine, total hip, and femoral neck of patients (BMD). At the 12th month fromRomosozumabAfter transitioning to denosumab,BMD Continues to Increase Until the 24th Month。
In another Phase III ARCH study, the subjects usedRomosozumabTreatment for 12 months, followed by 12 months of alendronate sodium treatment, at 24 monthsSignificantly reduced the incidence of new spinal fractures, with a median follow-up period of 33 monthsSignificantly reduced the risk of clinical fractures。
Compared with Alendronate Sodium,RomosozumabAt 12 monthsSignificantly increased BMD in the lumbar spine, total hip, and femoral neck. Compared with using alendronate sodium alone, usingRomosozumabTreatment for 12 months, followed by treatment with alendronate sodium for 12 months,Significantly increased BMD。

In October 2024, Zai Lab announcedPrimary data from the Phase III multicenter clinical study conducted in China to evaluate the safety and efficacy of KarXT. Consistent with previous global clinical studies, this registrational study met its primary endpoint, showing a significant difference compared to placebo at week five.KarXT’s PANSS total score decreased by 9.2 points, with statistically significant results.(-16.9 KarXT vs -7.7 placebo, p=0.0014)。
The study also met all key secondary efficacy endpoints, with scores on the PANSS Positive Symptoms Subscale, PANSS Negative Symptoms Subscale, PANSS Marder Negative Symptoms Factor, and Clinical Global Impression Scale for Severity showing improvement compared to placebo by the fifth week.(CGI-S)Scores and the Percentage of PANSS Responders at Week FiveAll showed significant improvement.. All key secondary endpoints were formally tested in a pre-specified order.
Compared with previous schizophrenia-related studies of KarXT, no new or unexpected safety signals were observed in this study. Treatment-related adverse events occurring at an incidence ≥10% in the treatment group and at least twice that of the placebo group included vomiting, tachycardia, nausea, systemic hypertension, dizziness, and diarrhea.

You Min Su isThe World's FirstApproved for use inType I Hypersensitivity Reaction(Including severe allergic reactions)Epinephrine Nasal SprayClinical trials have shown that its pharmacokinetic and pharmacodynamic characteristics are equivalent to those of injectable epinephrine, capable of rapidly alleviating allergic symptoms. Currently, the 2 mg dosage form(Applicable to weight ≥30 kg)Already inChina, the United States, and the European UnionApproved for marketing.

Trastuzumab deruxtecan is a DXd ADC targeting HER2 designed using Daiichi Sankyo's proprietary technology. In March 2019, Daiichi Sankyo and AstraZeneca reached a $6.9 billion collaboration to co-develop and commercialize the product globally.
Breast cancer is the main battlefield for Trastuzumab Deruxtecan.。HER2-Positive Breast Cancer(2L+)Following HER2-low breast cancer, this approval expands the indications for trastuzumab deruxtecan to earlier lines of therapy and includes patients with HER2 ultra-low expression.
The approval of this new indication is based on the Phase III clinical trial.Positive Results from DESTINY-Breast06.The StudyEvaluatedTrastuzumab DeruxtecanCompared with the chemotherapy selected by the investigatorHR-positive, HER2-lowOrUltra-low ExpressionAdvanced or Metastatic Breast CancerEfficacy and safety in patients.
Data show that, for HR-positive, HER2-low metastatic breast cancer patients who have not previously received chemotherapy, as reviewed by a blinded independent central review(BICR)Evaluation,Trastuzumab deruxtecan reduces the risk of disease progression or death by 38% compared to chemotherapy(n=713;HR=0.62;95% CI:0.52-0.75;p<0.0001). In median progression-free survival(PFS)Aspect,The treatment group with trastuzumab deruxtecan reached 13.2 months, while the chemotherapy group reached 8.1 months.
In HER2-low subjects, Confirmed Objective Response Rate in the Trastuzumab Deruxtecan Treatment Group(ORR)was 56.5%, higher than the chemotherapy group's 32.2%. In terms of median duration of response(DOR) FormulaIn terms of median progression-free survival, the trastuzumab deruxtecan group showed 14.1 months, compared to 8.6 months in the chemotherapy group.
In the HER2 ultra-low expression population(n=153;HR=0.78;95% CI:0.50-1.21), the median PFS in the trastuzumab deruxtecan treatment group was 13.2 months, compared to 8.3 months in the chemotherapy group. In confirmed cases ORRIn terms of efficacy, the trastuzumab deruxtecan treatment group reached 61.8%, higher than the chemotherapy group's 26.3%. The median...DOR In terms of duration, the trastuzumab deruxtecan treatment group showed 14.3 months, while the chemotherapy group showed 14.1 months.

This new indication approval is primarily based on a Phase IIIAPOLLO Clinical Research(ALTN-AK105-III-02)Achieved"Double Endpoint Positivity"Positive outcomes.
This is a multi-center, randomized, open-label, parallel-controlled Phase III clinical study aimed at evaluating Anlotinib combined with Penpulimab.Compared with SorafenibEfficacy and Safety of First-Line Treatment for Advanced Hepatocellular Carcinoma. The study enrolled a total of 649 patients with advanced liver cancer, of whom 40.9% had major vascular invasion and alpha-fetoprotein. (AFP)The proportion of patients with ≥400ng/mL was 49.2%.The primary endpoints of the study are OS and PFS.。
At the 2024 ESMO Congress,China Resources Tianqing announcedAPOLLO StudyClinical Outcomes。Data shows,Anlotinib Combined with Penpulimab GroupMedian PFS was 6.9 monthsThe control group was 2.8 months, with the risk of disease progression or death.Significantly reduced by 47%;Anlotinib Combined with Penpulimab GroupMedian OS was 16.5 monthsThe control group was 13.2 months, mortality riskSignificantly reduced by 31%。
Safety analysis showed that the safety data of anlotinib combined with penpulimab was consistent with the known risks, and no new safety signals were identified. The combination regimen of anlotinib and penpulimab is expected to provide a new option for first-line treatment in patients with advanced HCC.
On December 22, the official website of the National Medical Products Administration (NMPA) showed,Semaglutide Injection(Product name: NovoGain®)The marketing application for cardiovascular indications has been approved, applicable toReduce Major Adverse Cardiovascular Events in Adult Patients Diagnosed with Cardiovascular Disease and BMI≥27 kg/m²(Cardiovascular death, non-fatal myocardial infarction, non-fatal stroke)The Risk。
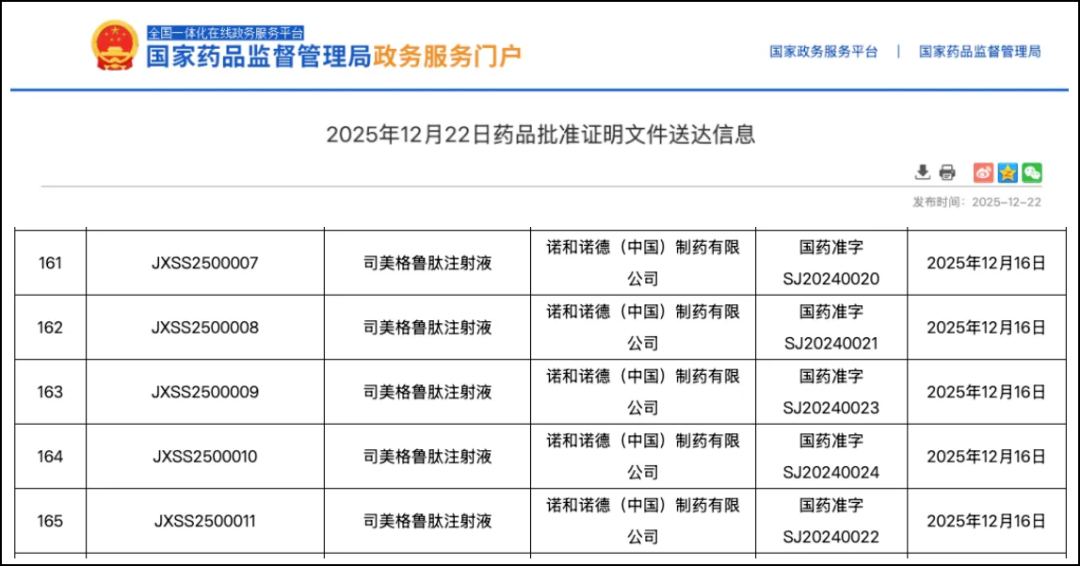
Source: NMPA official website
WorkOne of the important means in the standardized treatment of obesity.Semaglutide can achieve a weight loss of more than one-third of patients.20%, andCan reduce the risk of major adverse cardiovascular events by20%,Is the only one proven to both reduce weight and lower the risk of major adverse cardiovascular events(MACE)Weight-loss drugs。
The approval of this cardiovascular indication is based on the primary results of SELECT. As of now,The Largest Cardiovascular Outcomes Trial Completed for Obese Patients with Cardiovascular Disease, SELECT included a total of 17,604 overweight or obese subjects in41 Countries and regions800 Multiple research centers conducted studies.
The main results showed that, on the basis of standard treatment,Semaglutide Achieves 20% Reduction in MACE Risk Compared to Placebo, reducing the risk of cardiovascular death by 15%, the risk of heart failure composite endpoints by 18%, and the risk of all-cause mortality by 19%. Over a follow-up period of up to five years, MACE risk reduction was achieved regardless of the baseline age, gender, ethnicity, race, baseline BMI, or degree of renal impairment.
Moreover, a secondary analysis of the SELECT study data showed that: In the early stage of treatment, semaglutide has demonstrated cardiac protective effects prior to significant weight loss. This finding suggests that regardless of weight reduction, the use of semaglutide achieves the benefit of reducing MACE risk.
A Retrospective, Observational Real-World StudySTEER The study shows,Compared with tirzepatide, overweight patients continuously receiving semaglutide treatment/Patients with obesity and diagnosed cardiovascular disease have a significantly reduced risk of heart attack, stroke, and all-cause mortality.57%。At the same time,STEER The study results provide more evidence thatSemaglutideThe cardiac protective benefits demonstrated may be unique to the semaglutide molecule and therefore cannot be extended to others.GLP-1 OrGIP/GLP-1 Class of drugs。

MATINEE Study in 804 Cases with Evidence of Type 2 Inflammation(Eosinophil count ≥ 300 cells/μL)In patients with COPD, the efficacy and safety of mepolizumab were evaluated over 52–104 weeks. Enrolled patients included COPD clinical phenotypes with only chronic bronchitis, only emphysema, both conditions, or neither.
METREX trial in 836 patients(1:1 Randomization), to evaluate the efficacy and safety of Mepolizumab over 52 weeks, divided into eosinophilic phenotype groups(Eosinophil count at enrollment ≥150 cells/μL or ≥300 cells/μL in the past year)And non-eosinophilic phenotype group(Eosinophil count at enrollment <150 cells/μL and no evidence of ≥300 cells/μL in the past year)。
In the MATINEE and METREX trials, for patients with the eosinophilic phenotype, adding mepolizumab to standard triple inhaled therapy compared with placebo...Significantly reduce the annual incidence of moderate to severe acute exacerbations[MATINEE: Rate Ratio 0.79, 95% Confidence Interval [CI] 0.66, 0.94, p=0.01](Mepolizumab group AER=Exacerbation 0.80 times/year vs Placebo group=1.01 times/year)[METREX: Rate Ratio 0.82, 95% CI 0.68, 0.98, Adjusted p=0.04](Mepolizumab group AER=1.40 exacerbations/year vs placebo group=1.71 exacerbations/year).
In the pre-specified secondary endpoints of MATINEE, the Mepolizumab groupThe annual incidence rate of acute exacerbations leading to emergency visits and/or hospitalization was lower than that in the placebo group.[Rate Ratio 0.65, 95% CI (0.43-0.96) showed nominal significance after multiplicity adjustment](Mepolizumab group AER=Exacerbation 0.13 times/year vs Placebo group=0.20 times/year).
MepolizumabIs a targeted interleukin-5(IL-5)The monoclonal antibody was first approved by the FDA in November 2015. Since its approval, its sales have increased year by year. In 2024, the drug's sales have reached$2.279 billion, a year-on-year increase of 10%。
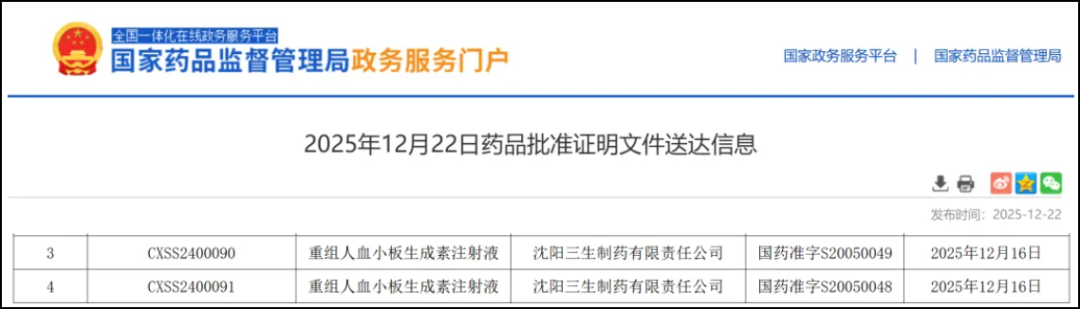
Tebiao is a recombinant human thrombopoietin independently developed by 3SBio.(rhTPO)Injection, which has the same amino acid sequence and pharmacological effects as endogenous thrombopoietin, primarily stimulates bone marrow megakaryocytes to promote the formation and release of platelets, thereby improving clinical symptoms caused by thrombocytopenia.
Insight database shows that the drugPreviously approved in ChinaThrombocytopenia After Chemotherapy for Adult Solid Tumors(CIT)、Adult Primary Immune Thrombocytopenia(ITP)AndPersistent or Chronic Primary Immune Thrombocytopenia in Children or Adolescents(ITP). Since the first approval,Tebiao's sales have been on the rise, reaching a total ofRMB 5.06 billion, an increase of 20.4% year-on-year.This is the fourth approved indication for the drug.
This timeThe approval of the CLDT indication is based on aMulticenter, Randomized, Double-blind, Parallel, Placebo-controlled Phase III Clinical Trial(CTR20230919)The positive results. The trial evaluatedTeBiAoEfficacy and Safety of Treatment Plans for Patients with Chronic Liver Disease-Related Thrombocytopenia Undergoing Invasive Surgery.
In July 2024, 3SBio announced that the clinical trial had reached its pre-specified primary endpoint,The primary efficacy endpoint was validation.TebeiaoIn patients with chronic liver disease-related thrombocytopenia scheduled for elective invasive surgery, maintain perioperative platelet count ≥50×10^9/L.More effective than placebo。
The research results show,Tepecta GroupProportion of subjects maintaining perioperative platelet count ≥50×10^9/L85.00%, the response rate in the control group was 12.50%, and the difference in response rate between the two groups was 67.90%.
The above results indicate that, compared with the control group,Tebiao GroupThe response rate significantly increased, and the primary efficacy endpoint reached a superiority conclusion. There was no significant difference in the incidence and severity of adverse events between the experimental group and the control group.
On December 22, the CDE official website showed that Dainippon SumitomoTotalDedabotumab for InjectionNew Indication Marketing Application Accepted. Based on clinical trial progress, the Insight database speculates that the indication is:First-line Treatment for Locally Recurrent Inoperable or Metastatic Triple-Negative Breast Cancer Not Applicable to PD-1/PD-L1 Inhibitors。

Dedabotumab, is aTROP2-targeted ADC, designed using Daiichi Sankyo's proprietary DXd ADC technology, consists of a humanized anti-TROP2 IgG1 monoclonal antibody(Developed in collaboration with Sapporo Medical University)Through a cleavable tetrapeptide linker with multiple topoisomerase I inhibitor payloads(A derivative of exatecan, DXd)Composed of connections. AstraZeneca holds the global development and commercialization rights for this product, excluding the Japanese market.
Currently, the product has been approved in China.HR-positive, HER2-negative breast cancerIndications.除此之外,DedabotumabIt has also been approved in the United States. EGFR Locally Advanced or Metastatic Non-Small Cell Lung Cancer with Mutations(NSCLC)Adult patients.
Source: Dingxiangyuan Insight Database
In October this year, Daiichi Sankyo announced the phase III TROPION-Breast02 data for datopotamab deruxtecan as a first-line treatment in patients with metastatic triple-negative breast cancer who are ineligible for immunotherapy. The results showed,Dedabotumab in Overall Survival(OS)And Progression-Free Survival(PFS)Statistically significant differences were observed in both primary endpoints, with clinically meaningful improvements achieved.
On December 24, the CDE website showed that Livzon Pharmaceutical's Class 1 new drugLecanemabThe listing application has been accepted. According to public information, Insight speculates that this drug was developed by Livzon Pharmaceutical.IL-17A/F Dual Target Inhibitor LZM012, The indication for this application may beModerate to Severe Chronic Plaque Psoriasis。
Screenshot source: CDE official website
LZM012 is a dual-target IL-17A/F inhibitor developed by Livzon Pharmaceutical. In July 2025, Livzon Pharmaceutical announced,LZM012 Phase III Clinical Trial for the Treatment of Moderate to Severe Plaque Psoriasis Successfully Meets Primary Endpoint。
This is a randomized, double-blind, positive-controlled multicenter Phase III study,Using a head-to-head study design, with the control group being secukinumab.(Cosentyx®). The primary endpoint of the study was the PASI 100 response rate at Week 12, and secondary endpoints included the PASI 100 response rate at Week 52, PASI 90 at Week 12, sPGA0/1 response rate, PASI 75 response rate at Week 4, etc.
Phase Ⅲ study data show that LZM012 has demonstrated multiple advantages, including:
Faster onset: LZM012 GroupThe PASI 75 response rate can reach 65.7% at Week 4 after a single dose at Week 0.In the secukinumab control group, W0/1/2/3 dosing was administered, and the PASI 75 response rate at Week 4 was 50.3%.LZM012 Achieves a Higher Response Rate Than Secukinumab with Just One Initial Injection`, reflecting its faster onset and superior efficacy.`
Excellent short-term efficacy: The PASI 100 response rate at Week 12 was 49.5% in the LZM012 group and 40.2% in the secukinumab control group, with the lower limit of the 95% confidence interval for the inter-group difference > -10% and > 0%, demonstrating the superiority of LZM012 over secukinumab; the PASI 90 and sPGA 0/1 response rates at Week 12 in the LZM012 group were > 80%, and the PASI 75 response rate was > 95%.
Long-Lasting StrengthUnder the two maintenance dosing regimens of LZM012 320 mg Q4W and 320 mg Q8W, psoriasis patients can continuously improve benefits, with PASI 100 response rates at week 52 being 75.9% and 62.6%, respectively, compared to 61.6% for Secukinumab 300 mg Q4W. This demonstrates that under the same four-week dosing cycle, the efficacy of LZM012 320 mg Q4W is significantly superior to Secukinumab.(75.9% vs 61.6%); whereas LZM012 320 mg Q8W, with a doubled dosing interval, demonstrates its PASI 100 response rate.(62.6%)Still superior to the control group's four-week dosing regimen(61.6%)。
High medication convenience: At Week 52, the PASI 100 response rate for LZM012 Q8W maintenance therapy was higher than that for secukinumab Q4W dosing after 12 weeks.
Excellent safety: Both short-term and long-term safety data show that the incidence of various adverse events in the LZM012 group was comparable to that in the control group, with a similar spectrum of adverse events.
On December 24, the CDE website showed that HutchmedFamritinib Tartrate TabletsThe marketing application has been accepted. Previously, the drug had been granted priority review by the CDE for the indication: treatment of patients with advanced, metastatic, or unresectable FGFR2 fusion or rearrangement as confirmed by testing who have received prior systemic therapy.Intrahepatic Cholangiocarcinoma(ICC)Treatment of adult patients.
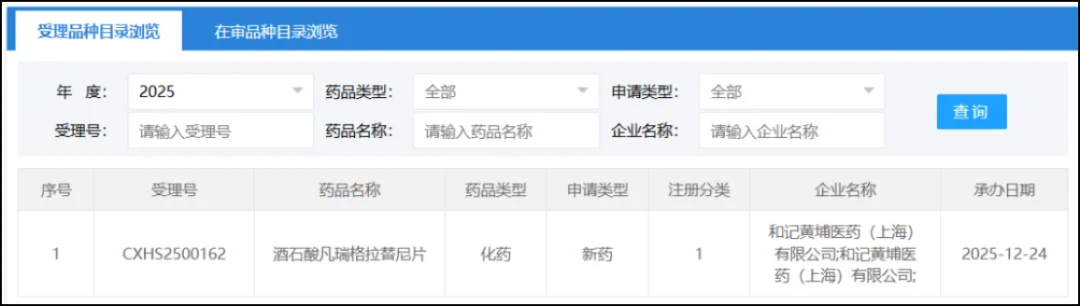
Screenshot source: CDE official website
According to the public data from Hutchmed, the Insight database speculates that Vanreglatib is the investigational Class 1 new drug Fanregratinib developed by Hutchmed.(HMPL-453)。
HMPL-453 is a novel, highly selective and potent FGFR 1, 2 and 3 inhibitor.Abnormal FGFR signaling has been found to contribute to tumor growth.(Through tissue growth and repair), promoting angiogenesis and inducing resistance to anti-tumor therapy. Abnormal FGFR gene alterations are considered driving factors for the proliferation of tumor cells in various solid tumors.
Hutchmed Plans to Develop HMPL-453 for TreatmentMesothelioma, Intrahepatic CholangiocarcinomaSolid tumors, etc.
In China, Hutchison MediPharma is conducting a single-arm, multi-center, open-label Phase II registrational study.(CTR20201011), aimed at evaluatingHMPL-453 for the treatment of patients with advanced intrahepatic cholangiocarcinoma harboring FGFR2 fusions/rearrangementsEfficacy, safety, and pharmacokinetics. The primary endpoint of the study is the objective response rate. (ORR) . Secondary endpoints include progression-free survival (PFS) , Disease Control Rate (DCR) , Duration of Relief(DoR) And Overall Survival (OS) 。
This Phase II trial has completed patient enrollment, and Hutchmed expects to release the clinical data of the study by the end of 2025. If positive results are achieved, the study will be used to support the submission of a new drug application for HMPL-453 to China's NMPA.
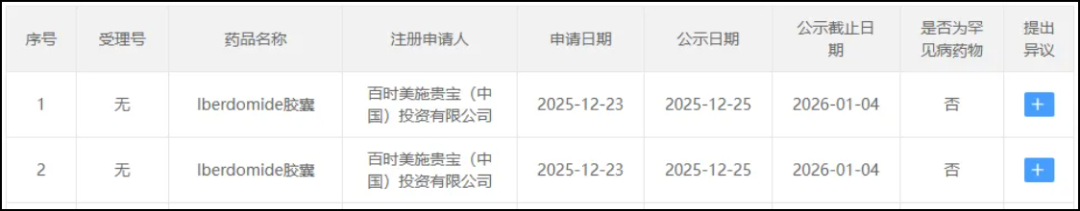
Screenshot source: CDE official website
Iberdomide is the first product of CELMoDCELMoD, as a new class of drugs, is expected to become a new cornerstone in the treatment of multiple myeloma when combined with other therapies.
In a Phase III, multicenter, two-stage, randomized, open-label studyEXCALIBER-RRMM(NCT04975997)ChinaResearchers evaluated Iberdomide in combination with Daratumumab and Dexamethasone(IberDd)Comparison of Daratumumab Combined with Bortezomib and Dexamethasone(DVd)In relapsed/refractory multiple myeloma(RRMM)Efficacy and safety in patients.
The study's dual primary endpoints include minimal residual disease(MRD)Negative Rate and Progression-Free Survival(PFS), other secondary endpoints include overall survival(OS), Objective Response Rate(ORR), Duration of Relief(DoR), Time to Disease Progression(TTP), Time to Next Line of Treatment(TTNT), and health-related quality of life(HR-QoL)。
The first phase of the study identified 1.0 mg Iberdomide as the optimal dose based on safety, pharmacokinetic, and efficacy data; the second phase enrolled approximately 664 patients, randomly assigned to either the IberDd or DVd treatment groups.
On December 25, the CDE website showed that Allist Pharmaceuticals' furmonertinib was proposed to be included in the breakthrough therapy category for a new indication, which is used forLocally Advanced or Metastatic Non-Small Cell Lung Cancer with EGFR PACC Mutation(NSCLC)First-line Treatment for Adult Patients。
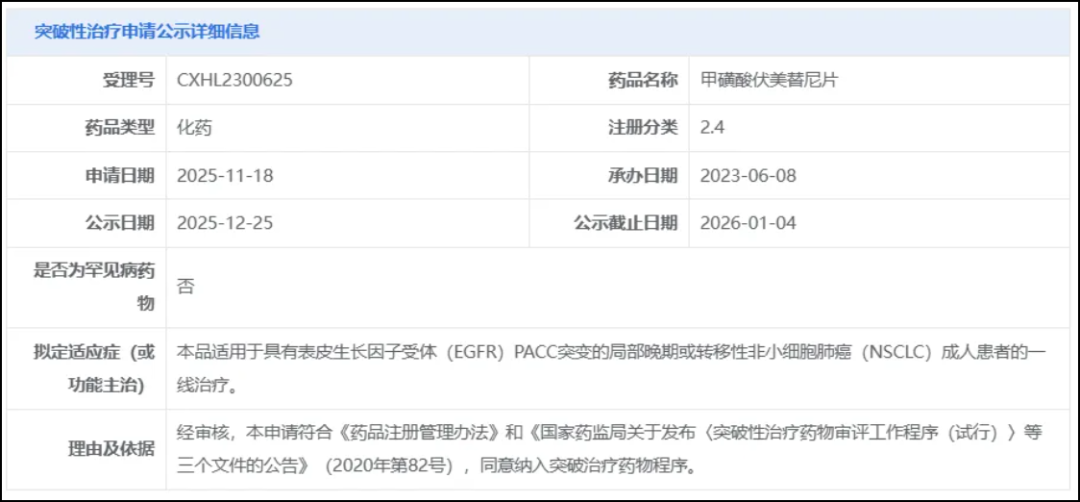
Screenshot source: CDE official website
Furmonertinib is a third-generation EGFR-TKI, which has previously been approved for two indications in China.Second-line and first-line treatment for adult patients with locally advanced or metastatic NSCLC with EGFR mutations. In July this year, the third indication application for Furmonertinib was accepted by the CDE and included in the priority review process for patients who experienced disease progression or were intolerant to platinum-based chemotherapy and haveEGFR Exon 20 Insertion MutationTreatment of adult patients with locally advanced or metastatic NSCLC.
The proposed breakthrough treatment this time is a new indication – forWith EGFR PACC mutationFirst-line treatment for adult patients with locally advanced or metastatic NSCLC.EGFR PACC mutations mainly include approximately 70 subtypes, accounting for about 12.5% of all EGFR-mutated NSCLC patients.Currently, there are no approved drugs for NSCLC patients with EGFR PACC mutations in China.
In the FURMO-002 study, the researchers evaluatedFurmonertinib First-Line TreatmentPreliminary Efficacy and Safety in NSCLC Patients Harboring EGFR PACC Mutations.AlisPoint out,FURMO-002 is the world's first prospective study conducted in the population with EGFR PACC multiple subtypes of advanced NSCLC.
As of June 2025, the Independent Review Committee(BICR)First-line treatment with 240 mg Furmonertinib in patients with advanced NSCLC carrying EGFR PACC mutations, evaluated based on RECIST v1.1Best Objective Response Rate(ORR)was 81.8%, the confirmed ORR was 68.2%, and the disease control rate(DCR)For 100%`, Median Duration of Response`(DOR)was 14.6 months, median progression-free survival(PFS)For 16.0 months. Additionally,Furmonertinib also showed good results in the study.Safety.
Furmonertinib is the main revenue source of Allist. Since its market launch, the drug's sales have increased year by year. In 2024, Furmonertinib achieved an annual revenue of 3.506 billion yuan, representing a year-on-year growth of 77.27%. In the first half of 2025, Furmonertinib continued to maintain a strong growth trend, generating product sales revenue of 2.36 billion yuan, with a year-on-year increase of nearly 51%.
On December 22, Grand Pharmaceutical announced that itsUsed forDiagnosing Prostate CancerInnovative radiopharmaceuticals under development (RDC) TLX591-CDx (Illuccix®, gallium Ga 68 PSMA-11) Phase III Clinical Trial Conducted in China, recentlyAchieved positive topline results, andSuccessfully achieved the primary clinical endpoint。

Source: Grand Pharmaceutical Official WeChat
This is a single-arm, open-label Phase III clinical study using TLX591-CDx and performing positron emission tomography/computed tomography in more than 100 patients with biochemical recurrence of prostate cancer. (PET/CT)or Positron Emission Tomography/Magnetic Resonance Imaging (PET/MRI) Testing to evaluate the diagnostic effectiveness of the product, while also assessing its safety and tolerability in the Chinese population.
Topline results from the clinical trial show,TLX591-CDx Overall Positive Predictive Value for Tumor Detection (PPV) Up to 94.8%(Confidence Interval, CI: 85.9%-98.2%). For recurrence in the prostatic bed area and metastasis to soft tissues, lymph nodes, and organs outside the pelvis(Non-bone metastasis)The tumor, PPV is 100.0%; for recurrence outside the prostatic bed area in the pelvic region(Including lymph nodes)For tumors, the PPV is 94.7%; for bone metastases, the PPV is 87.0%.
At the same time, the trial is based on different prostate-specific antigen levels among participants. (PSA) Grouped by baseline levels, TLX591-CDx demonstrated high PPV across all groups. Even in the subgroup of subjects with very low PSA levels, the PPV remained above 90%, suggesting that TLX591-CDx PET imaging detection is highly effective forEarly Diagnosis of Prostate Cancer Patients with Suspected Biochemical RecurrenceHas very positive clinical significance.
Moreover, more than two-thirds(67.2%)After PET imaging detection with TLX591-CDx, the treatment plan of the patient was adjusted compared to the initial plan at baseline. This indicates that PET imaging detection with TLX591-CDx has a significant impact on clinical decision-making.Optimizing Clinical Treatment Strategies for Prostate Cancer Patients with Suspected Biochemical Recurrence。
TLX591-CDx is aGlobal Innovation, Targeted Prostate Based on Radiopharmaceutical-Small Molecule Conjugation TechnologySpecific Membrane Antigen (PSMA) Diagnostic radiopharmaceutical, applicable for the diagnosis of newly diagnosed and recurrent prostate cancer. December 2021,TLX591-CDx Approved for marketing in the United States, expanded indications in March 2023, used for screening prostate cancer patients eligible for PSMA-targeted radionuclide therapy.


