Top Three Categories of FDA-Approved Digital Health Products (2016–2018): AI, Chronic Disease Management, and Telemedicine
Innovative design, intelligent R&D, and clinical validation are all critical steps in launching a medical product; however, without regulatory approval, many innovative products will never have the opportunity to reach patients and consumers.
In recent years, the rapid development of digital health has spawned numerous related products. In the United States, the Food and Drug Administration (FDA), the regulatory authority, has been continuously launching new initiatives and reforming existing regulations, aiming to streamline the approval process for new products and expand the digital health market.
VCBeat (WeChat: vcbeat) has reviewed the FDA’s regulatory history for digital health products. The main contents of this article include:
1. Inventory: FDA-Cleared Digital Health Products from 2016 to 2018
2. Review: How Does the FDA Regulate Digital Health Technologies and Products?
As of now, a total of 41 digital health products received FDA clearance in 2018.
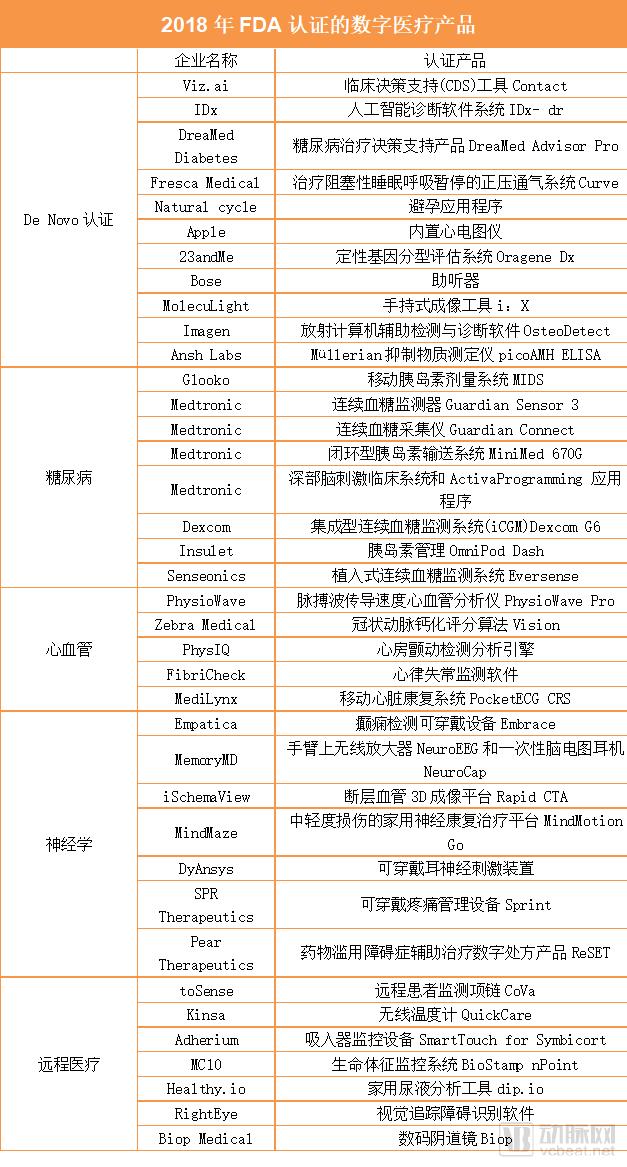
Category 1: De Novo Classification (Automated Class III Designation Review)
1. In February 2018, the FDA announced clearance for the marketing of Viz.ai’s Contact. Viz.ai’s Contact is a clinical decision support (CDS) tool designed to analyze CT scan results and determine whether a patient has suffered a stroke.
2. In April 2018, the FDA approved diagnostic company IDx’s De Novo application, allowing its artificial intelligence-based software system, IDx-DR, to enter the market for the automated detection of diabetic retinopathy in adult patients. This decision marked the first time the FDA had approved an AI-based diagnostic system for marketing in the United States that could provide diagnostic results without requiring interpretation by a clinician.
3. DreaMed Advisor Pro, a diabetes treatment decision support product launched by the Israeli company DreaMed Diabetes, has also received FDA De Novo clearance. Cloud-based DreaMed Advisor Pro is an artificial intelligence software that assists patients in managing type 1 diabetes. It analyzes data from continuous glucose monitors, insulin pumps, and self-monitoring devices to determine insulin delivery dosage.
4. Connected health company Fresca Medical has obtained FDA De Novo clearance to market its device, Curve, a positive airway pressure system for the treatment of obstructive sleep apnea. Curve comprises a flow generator, a lightweight ergonomic air delivery hose, and nasal pillows. Unlike other similar products, the device employs SmartValve technology, which uses less airflow than other systems and helps improve adherence among patients with sleep apnea syndrome.
5. The Natural Cycles contraceptive app has received FDA approval for sale to premenopausal women aged 18 and older. This algorithm-driven application, previously approved in the United Kingdom, is currently under review by multiple regulatory agencies. It helps users track their menstrual cycles and identify their most fertile days by measuring basal body temperature each morning and submitting information about their menstrual cycles.
6. Apple Inc. integrated an electrocardiogram (ECG) sensor into the fourth-generation Apple Watch, a feature that has received FDA clearance. Users require approximately 30 seconds to record an ECG, which is then stored in the Apple Health app. Through Apple Health Records, some users can also transmit this data directly to their physicians. Furthermore, the FDA has approved an Apple algorithm for detecting atrial fibrillation. In addition to on-demand ECG recordings, this functionality enables the watch to monitor heart rhythm in the background and issue notifications if irregularities are detected.
7. The 23andMe Personal Genome Service (PGS) is a qualitative genotyping assessment system applied to genomic DNA isolated from human saliva collected using the Oragene Dx OGD-500.001, enabling the simultaneous detection, reporting, and interpretation of genetic variants across a broad panel of polygenic tests. This assessment system is designed to provide users with direct access to genetic information that can facilitate discussions with healthcare professionals.
8. Bose hearing aids are designed to amplify sound for individuals aged 18 years or older with mild to moderate hearing impairment. Users can independently adjust the device according to their needs, without requiring pre-programming or hearing tests. The FDA classifies Bose hearing aids as Class II medical devices under the category of self-fitting air-conduction hearing aids, allowing them to be sold and used directly without the assistance of hearing healthcare professionals.
9. MolecuLight i:X is a handheld imaging device that enables clinicians to diagnose and treat skin wounds and is available for prescription use only. In accordance with FDA requirements, the MolecuLight i:X can be used to visualize and digitally record wound images, as well as to observe and digitally record fluorescence images generated by the wound when exposed to laser illumination.
10. OsteoDetect uses machine learning technology to analyze wrist X-rays, with the aim of identifying and highlighting distal radius fractures in adult wrists on posteroanterior (PA) and lateral (LAT) views obtained before and after review. The FDA considers OsteoDetect to be radiological computer-aided detection and diagnosis software and classifies it as a Class II medical device.
11. The picoAMH ELISA is an enzyme-linked immunosorbent assay (ELISA) intended for the quantitative measurement of anti-Müllerian hormone (AMH, also known as Müllerian inhibiting substance [MIS]) concentrations in human serum in vitro. AMH levels can significantly indicate menopausal status in women aged 42 to 62 years. The FDA requires that the picoAMH ELISA be used in conjunction with other clinical and laboratory findings; it should not be used alone to make diagnostic or treatment decisions. Furthermore, the FDA stipulates that the picoAMH ELISA is restricted to prescription use for in vitro diagnostic purposes only.
Category 2: Diabetes
12. Glooko, a digital health company specializing in diabetes care, has received FDA clearance for its Mobile Insulin Dosing System (MIDS). This app-driven tool directly adjusts insulin doses based on data collected from patients’ glucose meters. MIDS enables clinicians to create customized treatment plans and deliver them to patients with type 2 diabetes via the application.
13. Medtronic received four FDA approvals in 2018. The first approval expanded the indications for the Guardian Sensor 3, allowing patients to wear the sensor on the upper arm. This sensor is part of the MiniMed 670G system and is currently the only FDA-approved continuous glucose monitor that controls automated insulin delivery through a hybrid closed-loop system.
14. The second certified product is Guardian Connect. This device connects to smartphones and is designed for patients who require multiple daily insulin injections. The Guardian Connect system consists of the Guardian Sensor 3 and an associated transmitter, which sends continuously collected blood glucose data to the user’s smartphone via Bluetooth. The accompanying app can alert patients up to 60 minutes before a hyperglycemic or hypoglycemic event occurs.
15. The third approval extended the indication for the MiniMed 670G, a closed-loop insulin delivery system, to children aged 7 to 14 years with type 1 diabetes. In 2016, the MiniMed 670G became the first hybrid closed-loop insulin delivery system approved by the FDA; however, at that time, the FDA restricted its use to patients aged 14 years and older.
16. The fourth certified product is the Deep Brain Stimulation (DBS) Clinical System and the Activa Programming application. The DBS system is surgically implanted into the patient’s brain to deliver electrical stimulation to targeted areas. The Activa Programming application streamlines workflows and provides actionable information for neurologists and neurosurgeons.
17. Dexcom’s integrated continuous glucose monitoring (iCGM) system, the Dexcom G6, is the first approved interoperable continuous glucose monitoring system and the only iCGM recognized by the FDA that can integrate with other compatible medical devices and electronic interfaces, such as automated insulin delivery systems, insulin pumps, and blood glucose meters for diabetes management.
18. FDA Approves Insulet Corporation’s OmniPod Dash Insulin Management System for Market Entry. The product features a wearable insulin pump that assists patients in treatment by controlling basal rates and dosage. The Dash system also includes a handheld Personal Diabetes Manager with a touchscreen, which connects to the Pod via Bluetooth.
19. Senseonics’ implantable continuous glucose monitoring system, Eversense, has received FDA approval and will be launched in the United States. The company is already selling its second-generation device in Europe, the Middle East, and Africa.
Category III: Cardiovascular
20. PhysioWave Pro, a pulse wave velocity (PWV) cardiovascular analyzer launched by Stanford University-affiliated PhysioWave, has received FDA clearance. The device can monitor the blood vessels that transport blood from the heart to the body, as well as pulse and body weight, to assess whether users have cardiovascular disease.
21. Israeli deep learning startup Zebra Medical Vision has obtained FDA 510(k) clearance for its Coronary Calcium Scoring algorithm, which helps physicians quantify the extent of coronary artery calcification in patients using computed tomography (CT) scans.
22. PhysIQ’s atrial fibrillation detection and analysis engine has received FDA approval for use in patient care and clinical trials. The engine can be used in conjunction with PhysIQ’s other approved products to provide data analytics.
23. FibriCheck Announces FDA 510(k) Clearance for Its App, Which Uses Smartphone Cameras and Artificial Intelligence to Monitor Arrhythmias. The Company Expects to Launch Its Product in the U.S. Market in 2019.
24. MediLynx, a subsidiary of Medicalgorithmics, has obtained FDA 510(k) clearance for its mobile cardiac rehabilitation system, PocketECG CRS. Based on PocketECG technology, the product enables 30-day remote monitoring of patients with arrhythmia. The PocketECG CRS also features a built-in accelerometer and a touchscreen, facilitating data transmission to an online monitoring platform and a physician portal.
Category 4: Neurology
25. Empatica’s Embrace is a wearable device for seizure detection that received FDA 510(k) clearance this January. The product has been used in clinical trials by the pharmaceutical company Sunovion; however, users must obtain a prescription from a neurologist to access its seizure detection functionality.
26. MemoryMD’s two products, NeuroEEG and NeuroCap, have both received FDA clearance. NeuroEEG is a wireless amplifier worn on the arm that transmits electroencephalogram (EEG) signals to computers and cloud-based databases via Bluetooth. NeuroCap is a 19-channel disposable EEG headset, and the two products can be used together.
27. Rapid CTA, a product of the cerebrovascular imaging company iSchemaView, is a 3D imaging platform for computed tomography angiography (CTA). CTA scans help clinicians visualize patients’ cerebral arteries.
28. In June 2018, MindMaze received FDA clearance for MindMotion Go, a home-based neurorehabilitation platform designed for patients with mild to moderate impairments. Leveraging motion capture technology based on Microsoft Kinect, the product enables patients to engage in a series of activities within a 3D environment to facilitate recovery.
29. DyAnsys, a medical device company, has received FDA approval for its wearable auricular neurostimulation device to treat opioid withdrawal symptoms.
30. SPR Therapeutics has launched the Sprint Peripheral Nerve Stimulation (PNS) System, a wearable pain management device. The updated platform supports rechargeable batteries and includes a Bluetooth-enabled controller.
31. Pear Therapeutics’ product ReSET became the first FDA-approved digital therapeutic, indicated as an adjunct treatment for substance use disorders involving stimulants, cannabis, cocaine, or alcohol, to help patients regain control over addiction.
Category 5: Telemedicine
3. toSense’s CoVa monitoring system is a remote patient monitoring necklace that received its second FDA approval this year, allowing the company to update its software and add new features to the existing product, namely the measurement of stroke infarct volume and cardiac output. Additionally, the approval permits the device to connect to a mobile application that enables healthcare professionals to remotely view measurement data.
33. Connected health company Kinsa has launched a wireless device called Kinsa QuickCare. The QuickCare thermometer can read temperature in 8 seconds and retains most of the software features of its predecessor, the Smart Stick.
34-37. Smart inhaler company Adherium has obtained FDA 510(k) clearance to market its inhaler monitoring device for AstraZeneca’s Symbicort metered-dose inhaler—SmartTouch for Symbicort. The device can be attached to patients’ inhalers to monitor and improve medication adherence, helping clinicians identify when and how patients misuse their inhalers. Several months later, other Adherium products designed for ProAir HFA, Ventolin HFA, and Flovent HFA inhalers also received FDA over-the-counter authorization.
38. Sensor manufacturer MC10’s BioStamp nPoint system has received FDA 510(k) clearance for the first time. The system consists of reusable sensor patches that can monitor users 24 hours a day. The sensors can record various vital signs, such as movement and heart rate, and display the relevant data on a smartphone also provided by MC10.
39. dip.io, launched by the Israeli company Healthy.io, has also received FDA 510(k) clearance. This product is a home urine analysis tool that enables smartphone cameras to accurately read test strips without being affected by lighting or environmental conditions.
40. RightEye’s cloud-based eye-tracking system and software have received FDA 510(k) clearance. By recording, observing, and analyzing patients’ eye movements, the system and software help clinicians identify visual tracking disorders. Its key features include functional vision screening, reading assessment, sports vision assessment, training, and brain health.
41. Biop’s digital colposcope is designed for magnified visualization of the vagina, cervix, and external genitalia, enabling physicians to identify abnormalities such as lesions or cancer and to select biopsy sites. The Biop system consists of two main components: a digital colposcope unit connected to a main control unit. Connecting the Biop digital colposcope unit to the control unit allows for fully digital, high-resolution imaging of the cervix, with images viewable on a color touchscreen.
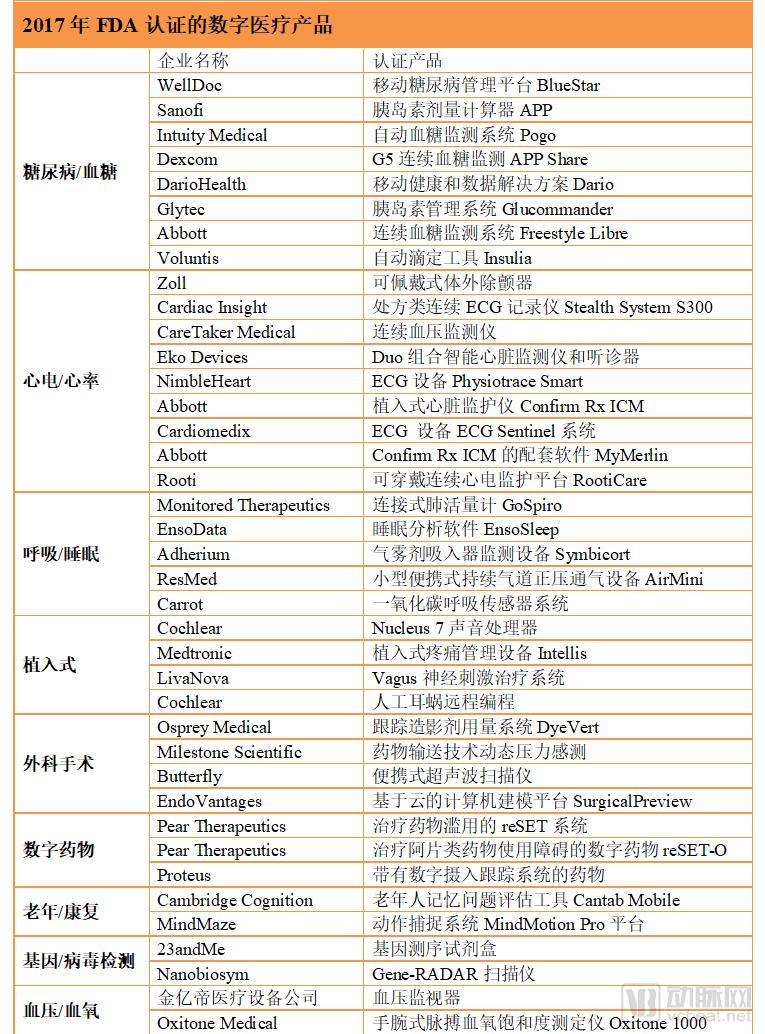
In 2017, a total of 43 innovative digital health products received FDA clearance, with diabetes management products and electrocardiogram (ECG) signal monitoring products accounting for a significant proportion.
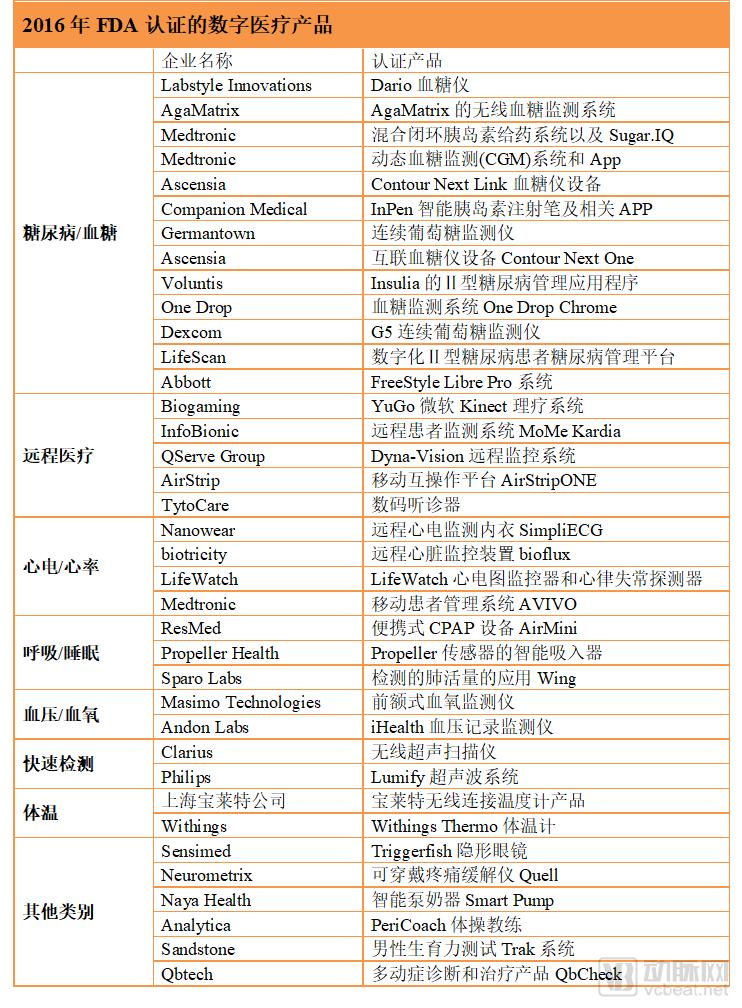
In 2016, a total of 37 digital health products received FDA clearance, with blood glucose/diabetes-related products being the most numerous.
In September 2013, the FDA officially released the “Mobile Medical Applications: Final Guidance,” marking the establishment of initial regulatory standards for the development of mobile health and laying the foundation for the subsequent growth of digital health.
According to FDA regulations, medical devices subject to regulatory oversight are classified into three categories:
1. Class I – Low risk, subject to general controls. Class I medical devices are medical devices that can connect to and control one or more medical devices, or display, store, analyze, or transmit specific patient data.
2. Class II – Moderate risk, subject to general controls and special controls. Class II medical devices can transform mobile devices into regulated mobile medical devices by leveraging accessories, displays, sensors, or functionalities of existing medically regulated devices.
3. Class III—High risk, subject to general controls and premarket approval. Class III medical devices can perform patient-specific data analysis and provide patient-specific diagnostic or treatment recommendations.
Following device classification, the corresponding applications must be submitted. The most common types of premarket applications include:
1. 510(k) (Premarket Notification);
2. PMA (Pre-Market Approval);
3. De Novo (Automated Class III Designation Review);
4. HDE (Humanitarian Device Exemption);
In July 2017, the FDA released the “Digital Health Innovation Action Plan” (DHIAP), which encompassed three key areas: issuing guidance for the implementation of new regulations, restructuring the regulatory framework for digital health products, and expanding specialized expertise. The plan outlined directives for ensuring high-quality, safe, and effective digital health solutions and declared its alignment with the objectives of the “Software as a Medical Device (SaMD) Guidance.”
By the end of 2017, the FDA had preliminarily completed several key initiatives under its “Digital Health Innovation Action Plan.” Among these were the draft guidance on “Clinical and Patient Decision Support Software,” the draft guidance on “Changes to Existing Medical Software Policies Resulting from Section 3060 of the 21st Century Cures Act,” and the final guidance on “Software as a Medical Device (SaMD): Clinical Evaluation.” These documents elaborate on certain important provisions of the 21st Century Cures Act and clarify the FDA’s role in the field of digital health.
In July 2017, the FDA also announced its decision regarding the Digital Health Software Pre-Certification (PreCert) Program. The FDA’s PreCert pilot program indicates that the agency will select and pre-certify specific digital health developers—such as companies evaluated against objective criteria, including excellence in software development/design practices and robust processes for validating software development and ensuring quality. Once pre-certified, low-risk mobile medical products developed by these digital health companies will be exempt from additional FDA review or subject to a streamlined review process.
In November 2018, the FDA announced reforms to the regulations governing 510(k) submissions. A 510(k) submission is a premarket notification filed with the FDA to demonstrate that the device seeking market clearance is as safe and effective as a legally marketed device that is not subject to premarket approval (PMA), i.e., it is substantially equivalent. The FDA announced that it would introduce a new regulatory pathway in 2019 to comprehensively replace the 510(k) process. For predicate devices, a 10-year time limit will be considered.
In December 2018, the FDA issued a new draft guidance on De Novo requests. In the United States, medical devices that lack a legally marketed predicate device cannot establish substantial equivalence through the 510(k) pathway to obtain marketing authorization, even if they are classified as low-to-moderate risk. To address such products, the FDA established the De Novo classification pathway several years ago, allowing them to be subject to general or special controls rather than being automatically classified as high-risk Class III devices. This approach reduces regulatory burdens on manufacturers and facilitates timely patient access to safe and effective medical devices.
The FDA’s newly released draft De Novo classification rule more transparently and effectively clarifies the documentation requirements for submissions under this pathway and the FDA’s review process. For instance, the proposed regulations and requirements will provide structure, clarity, and transparency to the reclassification process, including requirements related to the format and content of reclassification requests, as well as the processes and criteria for accepting, approving, denying, and withdrawing such requests. If finalized, this rule will facilitate the appropriate classification of novel medical devices, enabling device developers to leverage this pathway to bring more new devices to market.
References:
2.https://vcbeat.top/ZTMwMDEyZWI2MTAyNDg4N2E0OWNmOTgxNzczOTNhZmE=
3.https://vcbeat.top/MTkyMDA0MGFlZDVlMjA1ZDA3M2MzZmZjOGQzNTAzNTk=
4.http://www.appliedclinicaltrialsonline.com/look-fda-s-digital-health-action-plan