Sangamo Therapeutics Announces First-Ever In Vivo Gene Editing Milestone and FDA’s Accelerated Approval Pathway for Gene Therapy
Scientists had previously edited the human genome, but always in cells ex vivo. In February 2019, Sangamo Therapeutics, a company headquartered in California, announced the completion of the first in vivo gene editing in a clinical trial. Using zinc finger nucleases (ZFNs) as vectors, they inserted genes encoding functional enzymes into patients’ genomes to treat Hunter syndrome (mucopolysaccharidosis type II) or Hurler syndrome (mucopolysaccharidosis type I-H).
VCBeat’s New Medicine (biobeat1) believes that although the trial results are insufficient to prove therapeutic success, this milestone event still sends positive signals. Gene therapy will become a key development direction in the pharmaceutical sector from 2019 to 2022. The VCBeat New Medicine Database has also monitored 45 financing events involving global gene therapy companies from 2017 to the present. In 2018, the total amount of financing for gene therapy companies surged by 428.8% compared with 2017. Whether the gene therapy field in 2018 was merely a concentrated capital bet or the inaugural year for the industry’s takeoff remains unclear; nevertheless, its strong capital-attracting capability and future potential continue to make it one of the most noteworthy subsectors in the new medicine landscape.
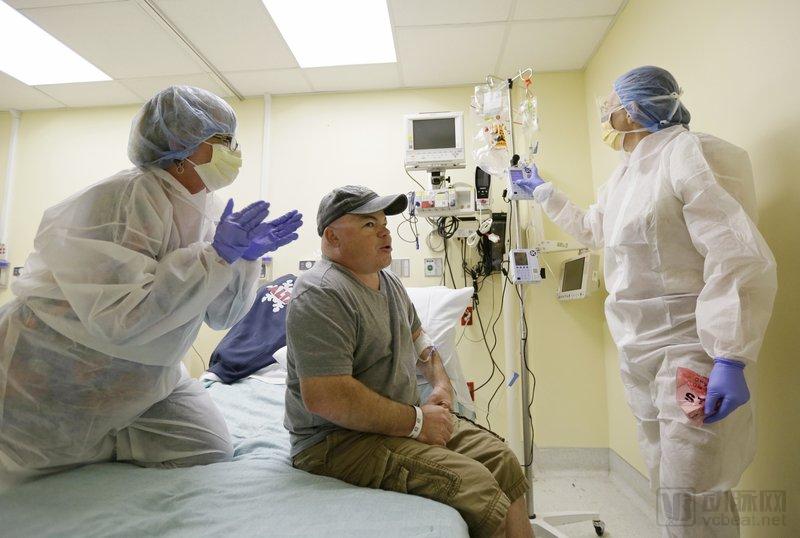
On November 13, 2017, Brian Madeux beganat UCSF Benioff Children's Hospital in Oakland, CaliforniaReceiving the first human gene-editing therapy for Hunter syndrome. On the left, his girlfriend, Marcie Humphrey, is applauding him. On the right is nurse Jacqueline Madden. On February 7, 2019, scientists announced a study involving the first use of gene editing in more than a dozen adults with metabolic disorders, including Madeux. (Image source: AP News)
Preliminary trial results indicate that two male patients with rare diseases now have very low levels of the corrected gene. In one patient with Hunter syndrome, levels of the deficient enzyme are approaching normal, but the immune system is affecting the treatment. In three patients with Hurler syndrome, enzyme levels have risen to normal. “This is a first step,” said Dr. Joseph Muenzer of the University of North Carolina at Chapel Hill, who participated in the clinical trial. “It just isn’t working strongly enough yet.”
Sangamo’s research involved male patients with Hunter or Hurler syndrome. In 2017, Brian Madeux of Arizona became the first person to undergo this experimental therapy. Via intravenous infusion, he received multiple copies of a corrected gene along with zinc-finger nuclease editing tools designed to insert them into his DNA. Results from him and seven other patients with Hunter or Hurler syndrome indicated that the treatment was safe, which was the primary objective of this early-phase gene therapy trial.
Gene therapy refers to the introduction of exogenous normal genes into target cells to correct or compensate for diseases caused by defective and abnormal genes, thereby achieving therapeutic objectives. Gene therapy technologies involve targeted gene delivery using viral vectors, non-viral vectors, and genetically modified cell carriers; they also encompass gene editing techniques such as CRISPR-Cas9, as well as CAR-T cell therapy.
The first human gene therapy approved by the U.S. National Institutes of Health (NIH) was conducted in May 1989. The experiment involved inserting a neomycin resistance gene into a retrovirus that had been rendered non-infectious but retained the ability to carry and activate the aforementioned gene. The virus was introduced into tumor-infiltrating lymphocytes (TILs) isolated from cancer patients, and the cells were then returned to the patients’ bodies. In 1999, Jesse Gelsinger, an American boy, participated in a gene therapy trial at the University of Pennsylvania. He died four days after treatment due to multi-organ failure caused by a severe immune response triggered by the viral vector. This incident marked a turning point in the development of gene therapy. In 2003, the U.S. Food and Drug Administration (FDA) temporarily halted all clinical trials using retroviruses to modify the genes of hematopoietic stem cells. However, after three months of rigorous review and deliberation, it permitted gene therapy clinical trials to resume.
Not long after the 2018 Christmas holiday, Scott Gottlieb, Commissioner of the U.S. Food and Drug Administration (FDA), together with Peter Marks, Director of the FDA’s Center for Biologics Evaluation and Research (CBER), jointly issued a statement on new policies to promote the development of safe and effective cell and gene therapies.
The advent of CRISPR/Cas9 gene-editing technology has broken through certain bottlenecks in gene therapy, improving both efficacy and safety, and ushering in a new wave of industry development. In the field of gene therapy, the U.S. National Institutes of Health (NIH) is playing a significant role. As of 2016, there were more than 2,300 clinical trials of approved gene therapies worldwide, with over half in Phase I clinical trials.
With the maturation of viral vector and gene editing technologies, gene therapy has transitioned from theory to reality. The market launch of three major gene therapy products—Kymriah, Yescarta, and Luxturna—in 2017 heralded the advent of the gene therapy era. Since 2013, companies engaged in gene therapy research and development have been highly sought after by European and American capital markets, securing substantial venture capital investment. Several enterprises, including Bluebird Bio, Celladon, uniQure, and Orchard Therapeutics, have completed initial public offerings (IPOs). Major domestic companies in China have also begun to make significant strategic investments in the field of gene therapy. For instance, BGI Genomics made a strategic investment in He Eye Specialist Hospital, initiating in-depth collaboration on gene therapy for hereditary retinal diseases associated with RPE65 gene mutations. Meanwhile, WuXi AppTec has expanded its layout of gene therapy clinical trials.
CBER has long been a relatively quiet division within the FDA. Its current high-profile emergence is indeed driven by the backlog of more than 800 Investigational New Drug (IND) applications for cell and gene therapies, which urgently require a safe and efficient pathway for review and approval.
The FDA projected that by 2020, it would receive more than 200 such investigational new drug (IND) applications annually; and by 2025, even based on current clinical success rates, the agency would approve 10–20 cell and gene therapy products per year. Gottlieb pointed out that the increased workload at the Center for Biologics Evaluation and Research (CBER) reflects a turning point in the development of cell and gene therapies and their application to human health, “similar to the late 1990s, when antibody drug development accelerated, and monoclonal antibodies have now become mainstream in modern therapeutic regimens.”
Gottlieb and Marks outlined three major initiatives in a joint statement. First, increasing staffing. The FDA is working to expand the staff of the Center for Biologics Evaluation and Research (CBER) review teams, with plans to add 50 clinical reviewers responsible for overseeing clinical investigations of cell and gene therapy products, keeping pace with the rapid expansion of new product development. Second, accelerating approvals. Emphasis was placed on utilizing the Regenerative Medicine Advanced Therapy (RMAT) designation, an expedited approval pathway introduced by the FDA in 2017 specifically for cell and gene therapies, which facilitates earlier and more frequent communication between applicants and the FDA. Third, issuing guidance. Gottlieb and Marks stated that the FDA would release a series of clinical guidance documents in 2019 related to various aspects of active product development, including guidance on the development of gene therapy products for specific diseases and guidance addressing manufacturing issues associated with gene therapies.
As innovative therapeutic solutions based on entirely novel mechanisms of action, cell and gene therapy products are ill-suited for the traditional approval framework. Therefore, to accelerate the regulatory review of these therapies, it is essential to address their fundamental characteristics and critical challenges.
When all or part of a gene is defective or missing at birth, or when genes change or mutate during adulthood, the way proteins are produced can be disrupted, potentially leading to health problems or disease. In gene therapy, scientists can choose to replace genes that cause medical issues, add genes to help the body fight or treat disease, or silence genes that cause problems.
To insert new genes directly into cells, specialized vectors are required to deliver the genes via genetic engineering. For example, viruses possess a natural ability to deliver genetic material into cells and can therefore be used as vectors. However, before viruses can be used to carry therapeutic genes into human cells, they must be modified to eliminate their capacity to cause infectious diseases. Currently, adenoviruses are commonly used vectors. The FDA considers the identification of safe and effective gene vectors to be key to making gene therapy products viable treatment options.
Before a company can market gene therapy products for human use, these products must undergo safety and efficacy testing so that FDA scientists can determine whether the therapeutic risks are acceptable in light of their benefits. Gene therapy holds promise for transforming medicine by creating new options for patients with conditions that are difficult or even impossible to cure. As scientists continue to make significant advances in this field, the FDA is committed to accelerating development by expediting the review of potentially life-saving breakthrough therapies.
Within the FDA’s regulatory framework, cell and gene therapies both fall under the jurisdiction of the Center for Biologics Evaluation and Research (CBER). CBER oversees blood products, vaccines, allergens, tissues, and cell and gene therapies, covering a narrower scope than the Center for Drug Evaluation and Research (CDER). VCBeat has compiled data on products approved by CBER since its establishment, revealing that its cumulative approval volume is even lower than the number of drugs CDER approves in just six months. Further analysis indicates that cell and gene therapy products do not yet constitute the mainstream of CBER’s approvals. Gene therapy products, in particular, remain a niche segment, accounting for only 18.35% of approvals, with merely three well-known products having reached the market to date. In light of the backlog of Investigational New Drug (IND) applications mentioned at the beginning of this article, it is evident that the path toward reforming CBER’s review process for cell and gene therapy products remains long and arduous.
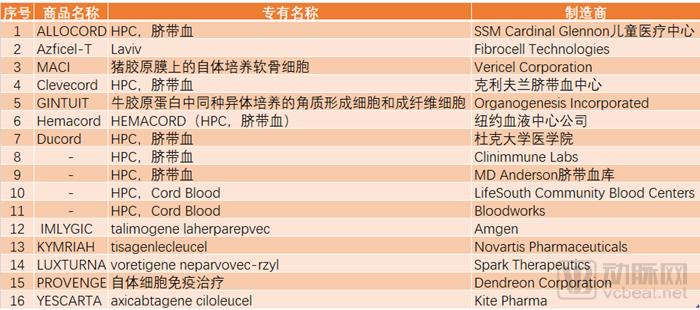
FDA-Approved Cell and Gene Therapy Products
Regenerative Medicine Advanced Therapy (RMAT), mentioned in the previous section, is a key focus of the FDA’s recent efforts to accelerate approval processes. On the FDA’s official website, the introduction to RMAT is placed under the category of “Cell and Gene Therapy Products,” highlighting its targeted nature and professional specificity. The legal basis for RMAT stems from the 21st Century Cures Act, signed into law by former U.S. President Barack Obama in late 2016.
In November 2017, the FDA released a draft framework for regenerative medicine, comprising two final guidance documents and two draft guidance documents. This framework clarifies the agency’s comprehensive policy approach to the development and oversight of novel cell therapies and other regenerative medicine products. It specifies that Regenerative Medicine Advanced Therapy (RMAT) designation is available for drugs (i.e., human drugs, including those regulated as biologics) that meet the definition of a regenerative medicine therapy. Such therapies are intended to treat, modify, reverse, or cure a serious or life-threatening disease or condition, with preliminary clinical evidence indicating the potential to address unmet medical needs for such diseases or conditions. Combination products may be eligible for RMAT designation when the biologic component contributes most significantly to the overall expected therapeutic effects of the combination product.
The FDA tends to draw parallels between Regenerative Medicine Advanced Therapy (RMAT) designation and Breakthrough Therapy designation, with the former resembling a Breakthrough Therapy designation tailored specifically for cell and gene therapy products. Furthermore, RMAT designation applications must be submitted concurrently with or shortly after an Investigational New Drug (IND) application, whereas Breakthrough Therapy designation can be requested after IND approval but prior to Phase II clinical trials. RMAT designation was introduced approximately five years later than Breakthrough Therapy designation. Currently, all three gene therapy products approved by the FDA (Kymriah, Yescarta, and Luxturna) accelerated their approval pathways through Breakthrough Therapy designation. The table below presents the status of RMAT designation applications accepted by the FDA during fiscal years 2017–2019.
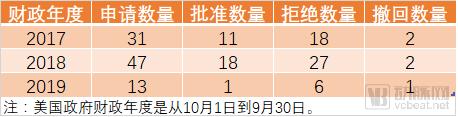
FDA’s Acceptance of RMAT Designation Applications in Fiscal Years 2017–2019
Here comes the highlight. The FDA has repeatedly emphasized in its statements that, due to the unique mechanism of action of gene therapy products, clinical trials of any scale cannot verify their sustained efficacy. In other words, conventional clinical trial endpoints are unlikely to be observed in patients enrolled in clinical trials of gene therapy products. The FDA’s solution is quite bold: it defers the evaluation of clinical trial endpoints for gene therapy products until after market approval, through Phase IV clinical trials, in exchange for advancing the timeline for product market approval as much as possible.
Certainly, the FDA would not dare to gamble with the lives and health of patients worldwide. On one hand, a hard threshold for gene therapy products to gain special access to clinical trials is that they must effectively treat serious diseases with no available alternatives. On the other hand, Gottlieb pointed out that a key current objective of the FDA is to establish a real-time monitoring system to precisely track safety issues after new products reach the market. Gottlieb emphasized, “Post-marketing clinical trials will become the key driver advancing the field of gene therapy.”
In fact, in addition to post-marketing clinical trials, the FDA can leverage various resources within the healthcare system, including insurance reimbursement. Taking Kymriah and Yescarta as examples, a prerequisite for Novartis and Gilead to secure reimbursement from the Centers for Medicare & Medicaid Services (CMS) was that “patients must demonstrate a therapeutic response within one month of treatment.” This represents the first “outcomes-based” reimbursement model pioneered in the United States. However, in real-world settings, many patients still show no response after three months of treatment.
Beyond high barriers to entry and stringent regulation, the development of clinical guideline documents mentioned by Gottlieb and Marks in their joint statement also constitutes a key component of the FDA’s comprehensive strategy. Institutionalizing processes is a preferred American approach to problem-solving.
As early as July 2018, the FDA issued six new guidance documents in the name of advancing the development of gene therapies, including three disease-specific guidances and three updates on gene therapy manufacturing.
Guidelines for the Treatment of Specific Diseases
Human Gene Therapy for Hemophilia.Gene therapy products for hemophilia currently under development are designed as one-time treatments that enable patients to sustain long-term production of the clotting factors that are missing or defective in their bodies, thereby reducing or eliminating the need for clotting factor replacement therapy. To define the appropriate development pathway for these products, the U.S. Food and Drug Administration (FDA) has released a draft guidance entitled “Draft Guidance on Gene Therapy Products for the Treatment of Hemophilia.” This new guidance aims to provide recommendations on clinical trial design and preclinical considerations to support the development of such gene therapy products. Among other elements, the draft guidance offers recommendations on the use of surrogate endpoints to facilitate accelerated approval of gene therapy products for the treatment of hemophilia.
Human Gene Therapy for Retinal Diseases.The treatment of retinal diseases is another hot area for gene therapy products. The FDA has issued the Guidance for Human Gene Therapy for Retinal Diseases. Currently, gene therapy products undergoing clinical trials for retinal diseases in the United States are typically administered via intravitreal or subretinal injection. In some cases, gene therapy products are encapsulated in devices intended for implantation into the eye. The new guidance focuses on specific issues related to gene therapy for retinal diseases and provides recommendations on product development, preclinical testing, and clinical trial design.
Human Gene Therapy for Rare Diseases.The National Institutes of Health reports that nearly 7,000 rare diseases affect more than 25 million Americans. Approximately 80% of rare diseases are caused by single-gene defects, and about half of all rare diseases affect children, indicating significant unmet medical needs. The Guidance on Human Gene Therapy for Rare Diseases provides recommendations on preclinical studies, manufacturing, and clinical trial design. This information is intended to assist sponsors in designing clinical development plans, addressing potential issues related to the limited size of study populations, feasibility, safety, and the interpretation of efficacy.
Updates to Guidelines for the Manufacture of Gene Therapies
"Chemistry, Manufacturing, and Controls (CMC) Information for Investigational New Drug Applications (INDs) in Human Gene Therapy Research", recommendations on how sponsors can provide sufficient CMC information to ensure the safety, uniformity, quality, purity, and potency of investigational gene therapy products. This guidance applies to human gene therapies, as well as products containing human gene therapies used in combination with drugs or devices.
“Testing Retroviral Vector Gene Therapy Products for Replication-Competent Retroviruses During Product Manufacturing and Patient Follow-Up”Provides additional recommendations for the appropriate testing of replication-competent retrovirus (RCR) during the manufacturing of gene therapy products based on retroviral vectors, as well as during the follow-up monitoring of patients receiving such products. Specifically, the guideline recommends determining the materials and sample sizes that require testing. The guideline also provides recommendations on general testing methods.
"Long-Term Follow-Up After Administration of Human Gene Therapy Products"》Provides recommendations on designing long-term follow-up (LTFU) observational studies to collect data on delayed adverse events following the administration of gene therapy products. Due to certain additional uncertainties inherent in novel technology platforms such as gene therapy—including issues related to the durability of therapeutic effects and the theoretical potential for off-target effects if gene insertion occurs incorrectly—focused long-term follow-up of patients in the post-marketing setting is required. This guideline describes product characteristics, patient-related factors, and preclinical and clinical data that should be considered when assessing the necessity for long-term follow-up, and also outlines the features of effective post-marketing surveillance.
In February 2019, Gottlieb issued a statement on a new initiative to promote the adoption of innovations in drug manufacturing. This initiative leverages collaborative efforts among regulatory agencies, industry, and academia by recognizing a series of agreed-upon “voluntary consensus standards” related to drug quality, which are nationally and internationally recognized pharmaceutical quality standards. Gottlieb noted that stakeholders and FDA staff would have the opportunity to propose drug quality standards for potential recognition by the FDA, thereby providing the industry with additional options for drug development and manufacturing. This move further extends the reach of clinical guideline documents.

Launch Details of the Three Blockbuster Gene Therapy Products
At the end of the article, VCBeat specifically compiled the launch details of three blockbuster gene therapy products—Kymriah, Yescarta, and Luxturna—and found that in an era when Regenerative Medicine Advanced Therapy (RMAT) designation was not yet widely utilized, Breakthrough Therapy Designation had effectively become the standard pathway for the rapid market approval of gene therapy products. In the United States, more than 800 gene therapy projects are still awaiting Investigational New Drug (IND) application approvals, and over 550 have entered clinical trials, with only a handful having received RMAT designation. In the future, securing RMAT designation may provide an additional safeguard for gene therapy products seeking to capitalize on emerging market opportunities.
Since the death of Jesse Gelsinger in the United States, the FDA has further strengthened regulatory oversight of gene therapy. Some clinical trials have been put on hold, as early-stage trials cannot guarantee consistent efficacy and safety. Despite these setbacks, technological advancements continue to drive progress in this field, and the future of gene therapy remains promising. Moving forward, VCBeat New Medicine will continue to analyze the development and R&D pipelines of gene therapy companies and produce the “2019 Gene Therapy Research Report” to assess whether new breakthroughs will emerge in the short term. We also welcome enterprises and experts in China currently conducting gene therapy-related research to contact us (WeChat: q19930797) to help refine this report.

Scan the QR code to follow the VCBeat New Medicine official account (biobeat1)