Beyond Amyloid: Rethinking Alzheimer's Therapeutics and Exploring Next-Generation Strategies
This article is republished from WuXi AppTec. VCBeat has been authorized to republish it.
Last week, Biogen and Eisai announced the early termination of the Phase 3 clinical trials of aducanumab, a beta-amyloid (Aβ) antibody developed by the companies for the treatment of patients with Alzheimer’s disease (AD). Since 2003, no new drugs have been approved in the AD field, making it a graveyard where numerous pharmaceutical R&D projects have failed.
In the wake of these disappointments, both academia and industry are reevaluating strategies for researching and treating Alzheimer’s disease (AD). The amyloid-beta hypothesis has been the most prominent and extensively tested theory explaining the etiology of AD over the past few decades. However, clinical therapies based on this hypothesis have consistently failed in clinical trials, including beta-secretase (BACE) inhibitors and anti-amyloid-beta antibodies.Although these therapies can successfully reduce Aβ levels or clear β-amyloid deposits in the brain, they do not help slow cognitive decline in patients.
Has the amyloid-beta hypothesis become outdated? What further efforts should be made in basic research to facilitate the development of more effective therapies? Beyond targeting Aβ, which other therapeutic approaches warrant attention?In this article, we will share our insights and information on this topic with readers, drawing from publicly available data and exclusive interviews conducted by WuXi AppTec with industry experts in Alzheimer’s disease (AD) drug development.
As multiple therapies targeting Aβ have failed in Phase 3 clinical trials, a commonly heard assertion is that we should seek treatment approaches beyond Aβ targeting. Does this mean the amyloid-beta hypothesis is obsolete? Nevertheless, many scientists remain firmly convinced of Aβ’s role in Alzheimer’s disease (AD), both in scientific research and drug development. What is the source of their confidence?
The β-Amyloid Hypothesis, originating in the 1980s, posits that an imbalance between the production and clearance of amyloid-β (Aβ) in the human brain leads to Aβ deposition. This triggers a cascade of events that ultimately results in neuronal damage and death, which is the fundamental cause of memory and cognitive decline in patients with Alzheimer’s disease (AD).
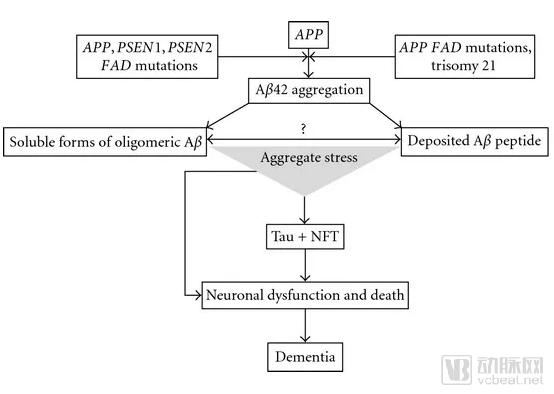
▲Illustration of the Amyloid-β Hypothesis (Image source: Reference [4])
The amyloid-beta hypothesis is currently the most scientifically supported explanation for the etiology of Alzheimer’s disease (AD). The strongest evidence comes from studies of patients with familial early-onset AD. These individuals develop AD before the age of 60 due to mutations in the APP, PSEN1, and PSEN2 genes. This genetic mechanism follows an autosomal dominant inheritance pattern, meaning that carrying just one copy of the mutated gene almost inevitably leads to AD. Among these three genes, APP encodes the precursor of Aβ, while PSEN1 and PSEN2 encode proteases responsible for cleaving the APP protein. These pathogenic mutations all result in an abnormally increased rate of Aβ production. Research on these mutated genes has facilitated the development of a series of mouse models of AD. Detection of Aβ deposition in the brains of patients also indicates that, in these early-onset AD cases, there is a very close correlation between the regions of Aβ deposition and the areas where subsequent neurodegenerative changes occur. Therefore, for patients with familial AD, the role of Aβ in the pathogenic mechanism is strongly supported by scientific evidence.
However, patients with Alzheimer’s disease (AD) caused by mutations in the APP, PSEN1, and PSEN2 genes account for only a small fraction of all AD cases. More than 95% of AD patients have late-onset AD, developing symptoms after the age of 65 and not carrying the gene mutations responsible for familial early-onset AD. Although the brain regions exhibiting amyloid-beta (Aβ) deposition are similar to those in early-onset AD patients, there is a substantial temporal gap between the onset of cognitive decline and the appearance of Aβ deposition in the brain.
Studies using positron emission tomography (PET) to investigate amyloid-beta (Aβ) deposition in the brains of patients with Alzheimer’s disease (AD) have shown that Aβ accumulation may begin up to 15 years before cognitive decline becomes apparent, and high levels of Aβ deposition are also found in many cognitively healthy older adults. In late-onset AD, the causes of Aβ deposition are less clear, and its association with cognitive decline is not as strong as it is in early-onset AD. For patients with late-onset AD, age is the greatest risk factor for developing the disease, followed by genetic variants in APOE and TREM2 as the most significant genetic contributors to increased risk. These risk factors show weak correlations with Aβ deposition; from this perspective, it is not surprising that multiple Aβ-targeting therapies have failed in patients with late-onset AD.
Does this mean that the “beta-amyloid” hypothesis is incorrect, or that researchers should abandon therapies targeting Aβ and shift their focus to exploring alternative treatments? Given the complexity of late-onset Alzheimer’s disease (AD) etiology, the scientific community and industry have already begun investigating other types of therapies, which is an essential path toward a more comprehensive approach to treating AD. However, therapies targeting Aβ should not be discarded. Studies of AD patient populations reveal that the impact of Aβ on AD pathogenesis exhibits a spectrum of varying magnitude across individuals. In patients with familial early-onset AD, Aβ plays a decisive role, whereas in those with late-onset AD, it may be merely one of many risk factors. Therefore, for patients in whom Aβ plays a predominant role in AD pathogenesis, Aβ-targeted therapies remain an effective treatment option.
This patient population may be the ideal candidates for testing targeted Aβ therapies, as they are typically younger and lack the complex confounding factors associated with aging that can affect trial assessments. Moreover, their disease progression is more predictable, allowing researchers to conduct interventional or preventive studies in advance. A well-known example in this regard is Roche’s trial evaluating the efficacy of crenezumab in a community in Colombia.
Crenezumab is a humanized monoclonal antibody targeting Aβ. In this Colombian community, some family members carry an inherited genetic mutation that typically causes them to develop Alzheimer’s disease (AD) around the age of 45. This clinical trial aims to initiate treatment five years before these patients exhibit AD symptoms. According to Dr. Mike Varney, Head of Research and Early Development at Genentech, who spoke at this year’s WuXi AppTec Global Forum, it may take more than a decade to obtain results from this trial.

▲ Dr. Mike Varney, Head of Research and Early Development at Genentech
This reflects, to some extent, the challenges faced in developing therapies for the treatment or prevention of Alzheimer’s disease (AD). Because AD progresses slowly, clinical trials using cognitive decline as the primary endpoint may require more than a decade to yield results. Furthermore, identifying patients with early-onset AD before symptom onset is not easy; apart from family medical history, genetic sequencing may be necessary to confirm their risk of developing the disease.
However, as the application of genomic sequencing expands, it will become increasingly easier to identify patients carrying specific genetic variants that play a key role in Aβ deposition during the pathogenesis of Alzheimer’s disease (AD). Furthermore, the globalization of AD patient advocacy organizations will help mobilize these patients, enabling them to contribute to the development and clinical trials of AD therapies. For investigational therapies targeting Aβ, their future success may hinge on stratifying the AD patient population to identify the subgroups most suitable for personalized treatment.
In the press release announcing Biogen’s discontinuation of aducanumab clinical trials, Mr. Michel Vounatsos, CEO of Biogen, stated: “This disappointing news once again underscores the complexity of treating Alzheimer’s disease.” Despite decades of scientific research on Alzheimer’s disease (AD), we have yet to fully elucidate its etiology and pathogenesis.
What breakthroughs are still needed in basic research for the scientific community?
Developing Animal and Cellular Models That Better Reflect the Pathogenesis of Alzheimer’s Disease
Recently, Nature launched a series on Alzheimer’s disease (AD), in which multiple experts in the field of AD research contributed articles providing comprehensive reviews of AD research from various perspectives. A major challenge in AD research lies in establishing experimental models that effectively mimic the pathogenesis of AD. Most current research models rely on mice. The advantages of murine models include their ease of genetic engineering and their convenient breeding and maintenance. However, wild-type mice do not naturally develop AD; therefore, gene mutations associated with familial AD must be introduced into mice to induce partial AD-like symptoms.
A review of the current status of Alzheimer’s disease (AD) mouse models indicates that early transgenic mouse models suffered from low reproducibility across studies due to random genomic insertion sites and variable levels of transgene expression. In recent years, mouse models generated using homologous recombination and gene-editing technologies have significantly improved the reproducibility of transgene expression in animal models. The authors of the review propose that all mouse models should be systematically evaluated to determine the extent to which they recapitulate the pathogenesis of AD, which will facilitate the selection of the most suitable animal models for AD research. They also propose a scoring system for evaluating AD animal models.
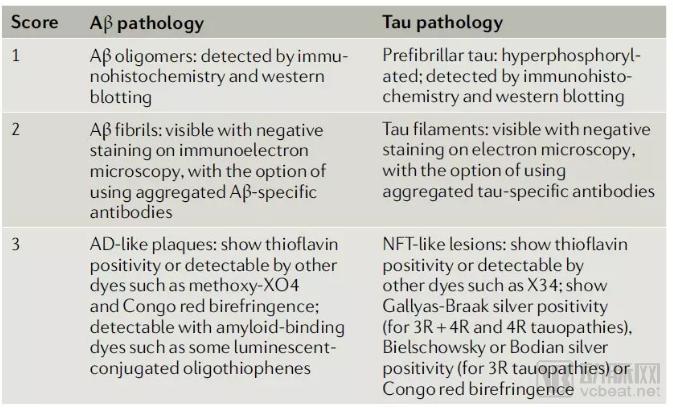
▲ A scoring system for evaluating whether mouse models can faithfully recapitulate the pathogenesis of AD (Image source: Reference [11])
One of the most significant challenges in studying Alzheimer’s disease (AD) using animal and in vitro models is how to simulate the impact of aging on AD. Many AD models recapitulate forms of the disease that more closely resemble familial early-onset AD rather than the late-onset AD seen in the majority of patients. From this perspective, while animal experiments can confirm the mechanistic role of amyloid-beta (Aβ), the findings do not necessarily translate into successful therapies. In the realm of animal studies, scientists have begun employing progeria animal models to explore the influence of aging on AD pathogenesis.
In terms of in vitro models, cell models generated using human induced pluripotent stem cells (iPSCs) and transdifferentiation may represent more accurate reflections of the genomic constitution and epigenetic characteristics of AD patients. iPSC technology enables the acquisition of cells from AD patients, which can be redifferentiated into neurons and glial cells in the brain, or even used to construct three-dimensional brain tissue and generate organoids in culture dishes. The advantage of this cell model is that it inherits all the genomic information of AD patients, facilitating the analysis of AD pathogenesis at the genetic level.
Transdifferentiation technology enables the direct conversion of fibroblasts or other cell types derived from patients into neurons or other cell lineages through treatment with chemical factors. The advantage of this technique lies in its preservation of the cells' epigenetic characteristics. Cellular aging is reflected not only in the accumulation of genomic mutations but also in epigenetic alterations, such as DNA methylation modifications. Models established using this technology retain the "traces" of aging embedded in the cells' epigenetic profiles, thereby helping scientists analyze the impact of aging on the pathogenesis of Alzheimer's disease (AD).
Imaging Techniques for Detecting AD Biomarkers
Non-invasive imaging techniques for detecting AD-related compound levels and brain function have advanced rapidly. Currently, PET technology enables the detection of Aβ and tau protein deposition in the brain. Furthermore, PET can assess cerebral glucose metabolism, which correlates with neuronal activity. Structural MRI allows for precise measurement of brain volume. These studies have delineated the various stages of onset and progression of Aβ and tau protein deposition, and correlated these changes with functional and structural alterations in the brain, thereby providing evidence to infer the roles of Aβ and tau proteins in the pathogenesis of AD.
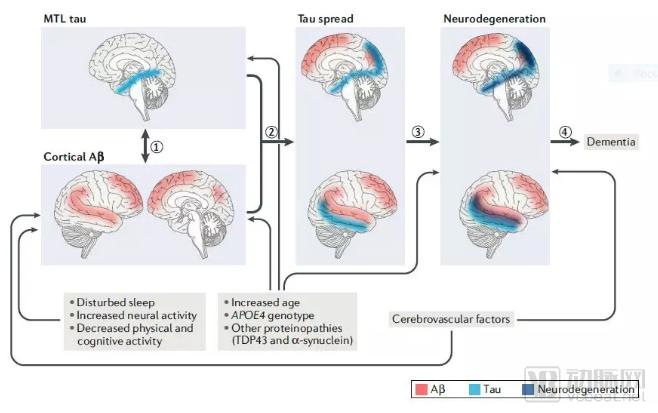
▲ AD pathogenesis proposed based on the detection of Aβ and tau protein deposition using imaging techniques (Image source: Reference [10])
However, the limitation of structural MRI and PET techniques for detecting metabolic levels is that these methods are not specific to particular cell types; the changes they detect represent a composite of alterations in both neurons and glial cells. Recent technological breakthroughs have begun to enable scientists to detect changes at the synaptic level.
Synaptic loss is a key hallmark of Alzheimer’s disease (AD) pathogenesis, and many investigational therapies are designed to prevent synaptic loss or enhance synaptic regeneration. The development of non-invasive imaging methods to assess the activity or quantity of brain synapses could provide researchers with a new clinical endpoint that does not rely on traditional cognitive assessments, thereby accelerating the clinical trial process for investigational AD therapies. Identifying biomarkers that can reliably predict cognitive changes in AD is crucial for both basic research and clinical development. According to the FDA’s clinical guidance on Alzheimer’s disease, if a biomarker can reasonably predict clinical benefit, the FDA may accept it as a surrogate endpoint to accelerate the approval of investigational AD therapies.
In addition to targeting Aβ, investigational therapies targeting other signaling pathways have been increasingly entering clinical trials in recent years. According to a 2018 report released by the organization “UsAgainstAlzheimer’s,” 16% of drugs in Phase II clinical trials target tau protein. Although the etiology of Alzheimer’s disease (AD) is complex and not yet fully elucidated, the ultimate decline in cognitive function is driven by synaptic loss and neurodegeneration. Therefore, targeting signaling pathways that affect synaptic function or neuronal survival may not only alleviate AD symptoms but also potentially offer therapeutic benefits for other neurodegenerative diseases.
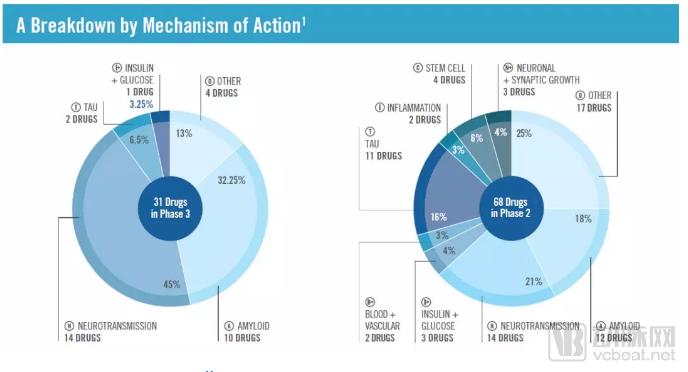
▲ Classification of Mechanisms of Action in the 2018 Alzheimer’s Disease Drug Development Pipeline (Image source: Reference [12])
Stimulating Neuronal Synaptic Regeneration
Loss of neural synapses is the biological mechanism mediating cognitive decline and memory loss in patients with Alzheimer’s disease (AD); therefore, preventing synaptic loss or enhancing synaptic regeneration has become a key research focus for many companies. For example, bryostatin, developed by Neurotrope, is a potent activator of PKCɛ that has been shown to restore synaptic density and promote synaptic maturation in animal studies. In a Phase 2 clinical trial involving 150 patients with late-stage AD, released earlier this year, this investigational therapy demonstrated improvements in patients’ cognitive performance.
NDX-1017, an investigational therapy from M3 Biotechnology, is an orally administered small-molecule compound capable of crossing the blood-brain barrier. It binds to hepatocyte growth factor (HGF) with high affinity, promoting HGF dimerization and activation of its receptor, c-Met. This mechanism stimulates neuritogenesis in neurons within the hippocampal region of the brain.
Targeting Neuroinflammatory Responses
Neuroinflammatory responses are not a disease feature unique to Alzheimer’s disease (AD). In many cases of chronic brain injury and other neurodegenerative diseases, chronic inflammatory responses are considered significant factors that exacerbate neuronal damage. AZTherapies’ investigational therapy, ALZT-OP1, is a combination treatment that shifts microglia in the brain from a pro-inflammatory state to a neuroprotective phagocytic state, thereby reducing brain inflammation while helping to clear amyloid-beta (Aβ) deposits. This therapy is currently being evaluated in Phase 3 clinical trials.
Denali’s investigational therapy, DNL747, is a small-molecule inhibitor targeting RIPK1 kinase. Enhanced RIPK1 activity in the brain drives neuroinflammation and necroptosis, thereby contributing to neurodegenerative diseases. This blood-brain barrier-penetrant small-molecule inhibitor has entered early-stage clinical trials.
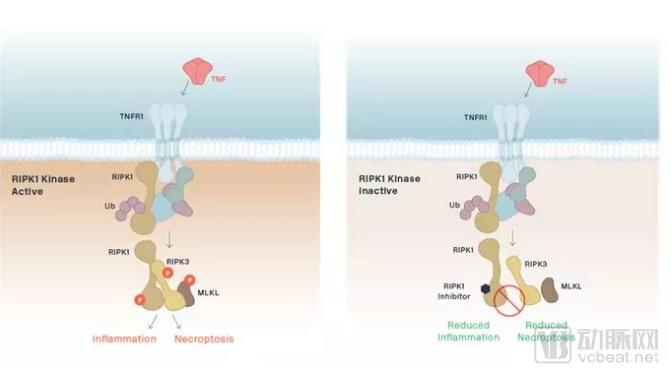
▲Schematic diagram of the mechanism of action of DNL747 (Image source: Denali Therapeutics official website)
Developing effective therapies for Alzheimer’s disease (AD) is not only a challenge for drug developers, but also for scientists engaged in basic research and regulatory authorities. We should pay tribute to the drug developers who have courageously pursued innovation, while also learning from past failures. Encouragingly, we are witnessing a diversification of AD drug development strategies. Through concerted multi-sector efforts, we look forward to the advent of effective AD treatments that will bring substantial improvements in quality of life for patients and their families.
References:
[1] Neuotrope. Retrieved March 22, 2019, from https://neurotrope.com/
[2] Mollinari et al., (2019). Transdifferentiation: a new promise for neurodegenerative diseases. Cell Death & Disease, https://doi.org/10.1038/s41419-018-0891-4
[3] Aber et al., (2017). Stem cell models of Alzheimer’s disease: progress and challenges. Alzheimer’s Research and Therapy. Doi: 10.1186/s13195-017-0268-4
[4] Reitz et al., (2012). Alzheimer's Disease and the Amyloid Cascade Hypothesis: A Critical Review. International Journal of Alzheimer’s Disease. Doi: 10.1155/2012/369808
[5] After the blowup of the biggest hope for Alzheimer’s, what’s next in the pipeline? Retrieved March 25, 2019, from https://www.statnews.com/2019/03/21/alzheimers-biogen-blowup-pipeline/
[6] Nelson et al., (2017). Bryostatin Effects on Cognitive Function and PKCɛ in Alzheimer’s Disease Phase IIa and Expanded Access Trials. Journal of Alzheimer’s Disease. Doi: 10.3233/JAD-170161
[7] M3 Biotechnology. Retrieved March 25, 2019, from https://m3bio.com/
[8] AZTherapies. Retrieved March 25, 2019, from https://aztherapies.com/
[9] Denali Therapeutics. Retrieved March 25, 2019, from https://denalitherapeutics.com
[10] Jagust (2018). Imaging the evolution and pathophysiology of Alzheimer disease. Nature ReviewsNeurosicence. https://doi.org/10.1038/s41583-018-0067-3
[11] Götz et al. (2018). Rodent models for Alzheimer disease. Nature Reviews Neuroscience. https://doi.org/10.1038/s41583-018-0054-8
[12] 2018 ALZHEIMER’S DRUG PIPELINE THE CURRENT STATE OF ALZHEIMER’S DRUG DEVELOPMENT. Retrieved December 4, 2018, from https://www.usagainstalzheimers.org/sites/default/files/2018_Alzheimers_Drug_Pipeline_The_Current_State_Of_Alzheimers_Drug_Development.pdf