Domestic Anti-Cancer Innovations Surge: Accelerated Approvals, Pharma Evolution, and Aggressive Capital
Editor’s Note: This article is republished from 21st Century Business Review, reported by Han Lu and edited by Chen Xiaoping. VCBeat has been authorized to republish it.
February 26, 2019, an ordinary Tuesday, became a remarkable day in the history of Chinese medicine due to a single prescription. For Chinese patients with melanoma, this day may well mark the dividing line between life and death.
At 4:00 p.m. that day, the first batch of toripalimab injection (brand name: Tuoyi) arrived at the pharmacy of Peking University Cancer Hospital, marking the official delivery and nationwide launch of the first domestically produced PD-1 monoclonal antibody anticancer drug in China.
For this milestone, Tuoyi embarked on a journey spanning more than three years: In December 2015, it received the first clinical trial approval for a domestic PD-1 monoclonal antibody in China; in March 2018, it became the first domestically produced PD-1 inhibitor to have its new drug registration application accepted; the following month, the drug registration application entered the priority review process; and in December, the product was approved for market launch. The approved indication was for unresectable or metastatic melanoma in patients who had failed prior systemic therapy. This pace saved at least half the time compared with previous approvals.
Melanoma is known as the "king of cancers." It is highly aggressive, has a high mortality rate, and is one of the most rapidly progressive, poorly prognostic, and refractory malignant tumors, with substantial treatment costs.
Junshi Biosciences, the manufacturer of Tuoyi, has priced the drug at RMB 7,200 per 240 mg vial (equivalent to RMB 30/mg), resulting in an annual treatment cost of RMB 187,200. In contrast, the imported product pembrolizumab, indicated for the same conditions, is priced at RMB 17,918 per 100 mg vial (equivalent to RMB 179/mg), with an annual treatment cost of RMB 609,212. The former amounts to only one-third of the latter (based on an average body weight of 60 kg), making it the lowest-priced drug in its class globally.
Patients eligible for the Beijing Bethune Public Welfare Foundation’s medication assistance program can receive four cycles of drug aid after completing four treatment cycles, reducing their annual out-of-pocket medication costs to approximately RMB 93,600. For patients who have long borne the burden of high-priced imported therapies or resorted to various informal channels to purchase medications abroad, the launch of Tuoyi is nothing short of a “miracle cure” descending upon them.
The approval of Tuoyi set a positive precedent, followed by a string of good news for domestically developed anti-cancer drugs.
In December 2018, Innovent Biologics’ PD-1 antibody drug, sintilimab injection, was approved for marketing in China for the treatment of relapsed or refractory classical Hodgkin’s lymphoma in patients who had undergone at least two lines of systemic chemotherapy. In January 2019, zanubrutinib, an investigational Bruton’s tyrosine kinase (BTK) inhibitor from BeiGene, received Breakthrough Therapy Designation from the U.S. FDA for the treatment of adult patients with mantle cell lymphoma (MCL) who had previously received at least one prior therapy. In February, Henlius (a subsidiary of Fosun Pharma) announced the approval of China’s first biosimilar, primarily indicated for the treatment of non-Hodgkin’s lymphoma.
Good news keeps coming. Under the spotlight of Sinopharm, a new landscape of interests is taking shape in the pharmaceutical market, and the “Chinese Drug God” is on the rise.
Domestic PD-1 Inhibitors Finally Hit the Market. The Excitement Is Shared Not Only by Patients but Also by Healthcare Professionals.
The first prescription for Tuoyi was issued one day earlier than expected. Industry peers and media outlets had been eagerly awaiting the moment it would be written on the morning of February 27, but Guo Jun, Deputy Director of Peking University Cancer Hospital and Secretary-General of the Chinese Society of Clinical Oncology (CSCO), could not wait. As a leading figure in melanoma treatment in China—affectionately known in the industry as “Boss Hei” due to his authoritative status—Guo also served as the principal investigator for the registrational clinical trials of Tuoyi.
On the 26th, Guo Jun had already left his office and was preparing to exit the hospital when he learned that the medication had arrived at the pharmacy. He immediately returned to his consultation room and issued the first prescription for Tuoyi. For patients with melanoma, every potential treatment option represents a lifeline.
In 1972, dacarbazine was approved by the FDA for the chemotherapy of melanoma. In the subsequent decades, only early-stage melanoma could be treated with local surgical resection, while other patients primarily received chemotherapy. However, data showed that the response rate to chemotherapy for melanoma was only 10–20%, with some studies reporting rates as low as 6–7%. Even when effective, chemotherapy could control tumor progression for only 1.7 months and failed to improve overall survival.
Twelve years ago, PD-1 inhibitors officially entered human clinical trials. Professor Lieping Chen, a leading authority in tumor immunotherapy at Johns Hopkins University in the United States, participated in organizing the world’s first clinical trial of a PD-1 inhibitor in cancer patients. The trial demonstrated the efficacy of PD-1 inhibitors against malignant melanoma, renal cell carcinoma, and lung cancer, ushering in a new dawn for the treatment of melanoma.
Li Ning, CEO of Junshi Biosciences, explained to a reporter from 21CBR that traditional chemotherapy and targeted therapy directly target tumor cells. “We refer to chemotherapy as cytotoxic therapy, which aims to kill tumors by poisoning cancer cells, whereas the therapeutic mechanism of PD-1 is entirely different.” PD-1 antibodies act on the human immune system by activating T lymphocytes suppressed by tumor cells, thereby restoring normal immune function.
“This approach enables a balance between the human immune system and tumors; in some cases, combination therapy can even completely eradicate tumors,” said Li Ning. In terms of therapeutic scope, no other anti-tumor drug currently matches the broad-spectrum activity of PD-1 inhibitors, with demonstrated efficacy across nearly all tumor types. “While conventional chemotherapy typically targets only one or two specific organs, PD-1 inhibitors exhibit systemic efficacy, showing significant advantages in both effectiveness and safety.”
According to data provided by Junshi Biosciences, among patients with unresectable or metastatic melanoma who had previously failed systemic therapy, treatment with Tuoyi (toripalimab) monotherapy resulted in tumor shrinkage of a certain magnitude sustained for a period of time in approximately 17.3% of patients. The disease control rate (DCR) was 57.5%, and the 1-year survival rate was 69.3%. The DCR of Tuoyi remained stable regardless of the number of prior lines of therapy or the melanoma subtype. Notably, for mucosal melanoma, which has a high incidence in China, early clinical data from a combination regimen of Tuoyi and an angiogenesis inhibitor showed a response rate exceeding 60%.
Behind these pale figures lie the hopes of individual lives saved.
Li Ning stated that, based on clinical data, the efficacy of domestically produced PD-1 inhibitors has at least matched that of imported drugs. “From a survival perspective, the median overall survival (OS) for melanoma patients treated with Tuoyi is approximately 24 months, whereas the median OS for those treated with Keytruda is less than 13 months. In terms of disease control rate (DCR), 60% of patients achieved disease control, with tumors either stabilizing or shrinking after treatment.” According to the prescribing information for Keytruda, its median OS is 12.1 months, and the DCR is 38.2%.
Such data has incentivized Junshi Biosciences to engage in clinical research for nearly all high-incidence malignant tumors in China. In addition to its existing indications, the company is simultaneously developing trials for more than ten tumor types, including nasopharyngeal carcinoma, gastric cancer, esophageal cancer, urothelial carcinoma, and lung cancer. The average total cost for Phase III clinical trials per tumor type is approximately RMB 200–300 million. Early clinical data indicate that domestically produced PD-1 inhibitors are effective and have the potential to become broad-spectrum anticancer drugs.
Junshi Biosciences is going all out to roll out its full portfolio, clearly aiming to capture market share as quickly as possible. For a cohort of innovative pharmaceutical companies in China, the window of opportunity has only just opened.
In 2009, China launched its second round of healthcare reform, and the IPO market reopened. That June, Chen Hai graduated from Peking Union Medical College Hospital, joined an investment bank, and became a pharmaceutical industry analyst. Over the past decade, he has served China’s first generation of pharmaceutical companies or their IPO projects, witnessing the rapid growth of their revenues and profits.
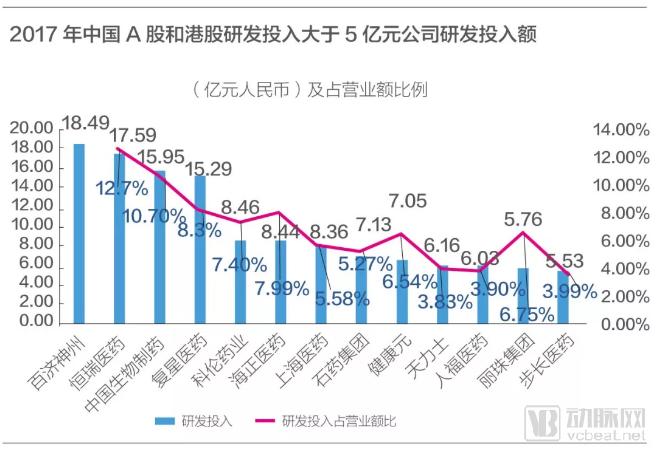
“An analysis of Chinese pharmaceutical companies’ product portfolios reveals that the market was once dominated by chemical generic drugs, with a scarcity of biotechnology firms and a lack of intellectual property (IP) and original proprietary rights,” said Chen Haigang. Over the past three to four decades, China’s pharmaceutical consumer market expanded rapidly, creating significant business dividends. A cohort of entrepreneurs secured their initial capital accumulation primarily through robust sales capabilities. In contrast, the independent research and development (R&D) of innovative drugs—often requiring ten years and costing up to $1 billion per new drug—did not guarantee market approval or commercial returns, making it an evidently unattractive proposition. Consequently, although China is home to more than 7,000 pharmaceutical companies, its R&D sector has long been characterized by large scale but limited strength.
“Even in drug development, many products have weak ties to scientific innovation, and the entrepreneurs behind them often lack a pharmaceutical background. The first generation of pharmaceutical companies was predominantly sales-driven,” Chen Haigang told 21CBR.
In 2015, Chen Haigang left the secondary market and joined Xingze Capital to engage in healthcare investment, marking a new starting point in his career. That year, China’s pharmaceutical industry reached a watershed moment.
“Around 2015, China saw its first wave of biotechnology companies, with startups focused on antibodies, CAR-T, and other areas emerging in large numbers. Compared to the first generation, this cohort of innovative pharmaceutical companies demonstrated significant iteration, creating a generational gap.” Chen Haigang clearly perceived that many R&D-driven companies had sprung up, featuring products with well-defined mechanisms of action and genuine clinical value, while their founding teams were predominantly composed of scientists.
Waves Rise from Gentle Ripples: The Ascendancy of This New Force in Pharmaceuticals Is Propelled by Multiple Forces.
First, there is the human factor.
The biopharmaceutical sector is an arena for elite entrepreneurship; success does not come merely from having a keen market sense or understanding consumer demand, but often requires extensive scientific training.
A closer look at the founders of China’s innovative pharmaceutical companies, categorized by their establishment dates, reveals uniformly impressive credentials: Xiaodong Wang, founder and Chair of the Scientific Advisory Committee of BeiGene, is a member of the U.S. National Academy of Sciences, a foreign associate of the Chinese Academy of Sciences, and Director of the National Institute of Biological Sciences, Beijing; Jinzi Wu, founder of Ascletis Pharma, previously served as Vice President of the Executive HIV Drug Discovery Department at GlaxoSmithKline in the United States; Ying Du, founder of Zai Lab, was formerly the founder of Hutchison MediPharma...
Among them, many were part of the first wave of Chinese students who studied abroad in the 1980s. Having gained extensive experience, knowledge, and expertise through overseas study and work, they have accumulated the capital and confidence necessary for entrepreneurship. After 2017, another wave of senior executives from multinational corporations returned to join local innovative enterprises.
In March 2017, Wang Liqun joined Fosun Kite as CEO, having previously served for many years at Bristol-Myers Squibb, AstraZeneca, and the GlaxoSmithKline China R&D Center. In January 2018, Li Ning left his position as Vice President at Sanofi to join Junshi Biosciences. Four months later, Wu Xiaobin, then Country Manager of Pfizer China and President of Pfizer Essential Health Greater China, announced his departure; subsequently, Beigene officially appointed him as General Manager and President for China. During the same period, Zai Lab announced that Liang Yi, former Vice President of AstraZeneca China and Head of Oncology Business, had joined the company as Chief Commercial Officer and President of Greater China.
Wu Xiaobin recalled that when he first came into contact with BeiGene in 2014, he was impressed by the company, as developing innovative drugs in China felt like “a distant prospect” at the time. It was only after a colleague left to join BeiGene that he began to reconsider this view. Around 2017, six or seven domestic innovative pharmaceutical companies extended offers to Liang Yi, who ultimately chose Zai Lab. He first heard of Zai Lab in 2015, while overseeing AstraZeneca’s oncology business: “It was quite coincidental; I was already tracking the product pipeline that Zai Lab had licensed in, which is how I learned about this domestic company.”
Meanwhile, professional investors have also begun to emerge.
Chen Haigang frankly stated that in pharmaceutical investment, capital contribution is only one aspect; what is more critical is making the right judgments. If the initial project selection errs in choosing the drug target, no amount of funding will suffice. “While the internet sector relies on traffic or operational data metrics, for biotechnology companies, investors need to understand what the company is doing, including the selected targets and their corresponding mechanisms of action. This all takes time.”
In the United States, Apple Inc. and Genentech (the world’s first biotechnology company) were both founded in 1976, with venture capital investment in IT and biotechnology taking off almost simultaneously. In China, however, venture capital in the biotechnology sector lagged by at least a decade but has begun to gain momentum in recent years.
In addition, the regulatory environment has undergone drastic changes. In 2015, the China Food and Drug Administration (CFDA), which has since been reorganized into the National Medical Products Administration (NMPA), launched a series of reforms in China’s pharmaceutical policies. The two most core initiatives were: first, to conduct self-inspections of clinical trials and initiate the consistency evaluation of generic drugs. Approved generic drugs are subject to phased quality consistency evaluations based on the principle of maintaining consistent quality and therapeutic efficacy with original reference listed drugs, requiring generics to achieve equivalent quality and efficacy; second, to improve efficiency across all stages of new drug approval, allowing direct submission for approval if overseas multi-center clinical trials include sufficient Chinese data.
This proactive drive for change stems from profound real-world pressures: China’s aging population is intensifying, the incidence and absolute numbers of diseases—particularly malignant tumors—are rising year after year, and public health expenditures continue to increase. These trends necessitate that the drug review and approval system keep pace with the times.
Under the former *Measures for the Administration of Drug Registration*, the standards for bioequivalence studies of generic drugs were unclear, and there were numerous loopholes in the submission and approval process.
On one hand, the quality of generic drugs fails to meet standards; on the other, the approval process for new drug launches is lagging. According to the regulations of the China Food and Drug Administration (CFDA), innovative drugs entering the Chinese market were required to submit clinical trial data from Chinese subjects, a measure intended to ensure efficacy and safety. However, as clinical trials typically take 1–3 years to complete, coupled with backlog in review processes, the domestic launch of innovative drugs has historically lagged behind overseas markets by more than five years, while independently developed drugs often require over ten years to reach the market.
Before returning to China, Li Ning held various positions at the U.S. FDA, including reviewer, senior reviewer, review team leader, and division director. He observed that the slow pace of new drug approval in China was also constrained by a shortage of reviewers. “With a population of 300 million, the United States has thousands of reviewers, whereas China, with a population of 1.4 billion, has only slightly more than 100 technical reviewers. As a result, it takes two years just to obtain approval for a clinical trial.”
The implementation of the new policy serves as a precise correction of interests, akin to walking a tightrope. It aims to restore the proper role of generic drugs, drive down prices of originator drugs after patent expiration, accelerate the market entry of new drugs, and ultimately reduce healthcare costs across society. As Wu Zhen, then Deputy Director of the China Food and Drug Administration (CFDA), stated at the time, “As long as there is clinical need and public benefit, the earlier the availability, the greater the benefit to the public.” In 2017, the CFDA officially joined the International Council for Harmonisation of Technical Requirements for Pharmaceuticals for Human Use (ICH), aligning drug approval processes with international standards and significantly narrowing the gap. Currently, clinical trial approvals can typically be completed within three months.
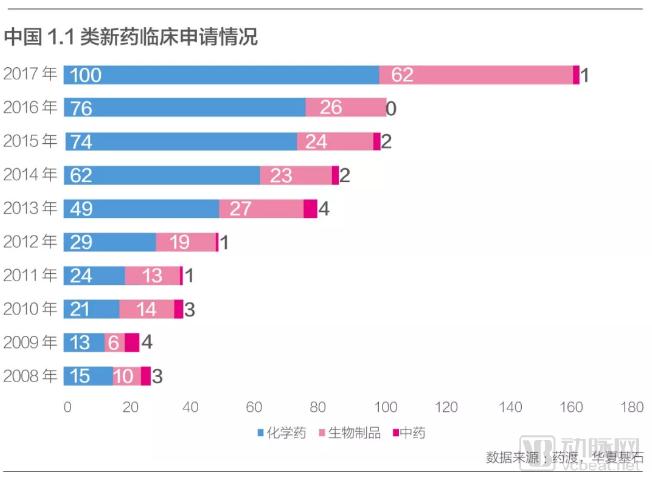
From 2008 to 2018, China approved a total of 38 Class 1 innovative drugs, with nine approvals in 2018 alone, accounting for nearly one-quarter of the total. In that year, China approved the marketing of roxadustat (brand name: Evrenzo), a “first-in-class” new drug developed by FibroGen and AstraZeneca for the treatment of renal anemia, which had not yet been launched in any other country. This marked the first time that China surpassed foreign markets by achieving the global first launch of a new drug.
New Drug Development: The Thin Line Between Pioneers and MartyrsIt is widely rumored in the industry that an innovative pharmaceutical company, now publicly listed, had only tens of thousands of yuan left in its accounts by the end of 2013. Several rounds of visiting investors showed no interest in investing. A group of founding employees, unable to endure the hardship, chose to resign. Out of consideration for their shared struggles, the company retained their original shareholdings. Unexpectedly, several years later, when the company went public through an IPO, these individuals saw their net worth double unexpectedly.
New drug research requires not only technology but also a business model.
Zai Lab, founded in 2014, is positioned as an innovative biopharmaceutical company focused on the research and development of therapies for cancer, autoimmune diseases, and infectious diseases. From its inception, the company adopted a “dual-engine” strategy combining in-house R&D with external licensing. It subsequently established two manufacturing facilities in Suzhou to enable local production of its future marketed drugs and built a nationwide commercialization team.
Liang Yi candidly acknowledged that even if Zai Lab began developing new drugs in 2015, given a 10- to 15-year development cycle, results would likely not be seen until 2030. The current R&D collaboration model, however, enables accelerated progress by “standing on the shoulders of giants.” “We are not simply engaging in license-in arrangements to introduce products for sale in China; rather, we are jointly completing development in China and achieving commercialization.”
In December 2018, Zai Lab’s first commercialized product, Zejula (niraparib), was launched. It is the first PARP inhibitor approved in Hong Kong, China, for maintenance treatment of all patients with platinum-sensitive recurrent ovarian cancer, regardless of BRCA mutation status. The product had previously been approved in the United States and Europe. This initial step into commercialization provided positive cash flow to support further R&D and innovation.
Throughout the journey to bring the PD-1 inhibitor to market, clinical expenses totaled approximately RMB 1.5 billion. Li Ning lamented that the company had been grappling with anxiety stemming from uncertainty, making life-or-death decisions at every turn. “Previously, while managing operations in more than 20 countries for a multinational corporation, I faced challenges, but they were worlds apart from the current race against time where survival hangs in the balance.”
Fortunately, top-level design has provided a boost to pharmaceutical R&D. In 2018, the National Healthcare Security Administration decided to launch the pilot program of volume-based procurement in “4+7” cities, completing drug procurement by specifying purchase volumes and awarding contracts to the lowest bidders. This move effectively pressured innovation from the payment end.
In simple terms, pricing and volume-based tendering are conducted among generic drug varieties that have passed the consistency evaluation. The 11 pilot cities allocate 60%–70% of the market share to the winning bidders, who exchange volume for lower prices, while other companies can only share the remaining 30%–40%. In the short term, pharmaceutical companies’ sales profits will inevitably be affected. In the long run, the valuations of generic drug manufacturers will quickly hit a ceiling, because once more than three companies pass the consistency evaluation for a given variety, price competition becomes intense, forcing generic drug manufacturers to survive by offering a diverse portfolio of products at low costs.
Policy implementation quickly transmitted to the capital markets. In September 2018, as soon as the symposium on the new policies concluded, pharmaceutical stocks in both the A-share and Hong Kong stock markets plummeted. Particularly in the Hong Kong market, 14 pharmaceutical stocks lost hundreds of billions of yuan in value in a single day.
On January 3, 2019, the market capitalization of China Biologic Products (01177.HK) fell to HK$58.369 billion, while that of BeiGene (06160.HK) stood at HK$62.508 billion on the same day. A pharmaceutical company with an annual profit of HK$2.1 billion surprisingly experienced a market cap inversion with a peer that continued to post losses. Some view this day as a watershed moment in China’s era of pharmaceutical innovation, marking the beginning of a reshuffling among pharmaceutical giants.
Amidst the vast prospects, talent is accelerating its convergence, and capital is becoming more active, willing to bear high risks.
After joining BeiGene in 2018, Wu Xiaobin found that the company’s internal scientists were thoroughly versed in new targets, scientific literature, and the pros and cons of various treatment modalities. The clinical trial capabilities were not only fast and efficient but also conducted across multiple global centers, to the extent that “in many areas, I became the one who needed to learn.”
“At Junshi Biosciences, we’ve heard the word ‘first’ too many times: the first to receive clinical trial approval for a PD-1 inhibitor, the first PD-1 inhibitor approved for market launch... But on the flip side, there is a lack of experience,” Li Ning admitted. From a corporate perspective, every step has been a journey from zero to one. However, most team members come from domestic or international innovative pharmaceutical companies and have witnessed the birth of many “first-in-class” oncology drugs; their individual expertise can fill the company’s experiential gaps. For instance, drug registration submissions require an international format, a task few people in China have ever performed. Yet many employees at Junshi, including Li Ning himself who has worked at the FDA for many years, are familiar with this process. They know how to present clinical study data to help regulatory authorities better understand it, thereby avoiding many detours.
After joining Zai Lab, Liang Yi immediately established commercialization teams for mainland China, Hong Kong, and Macao. The team-building effort in Hong Kong was particularly remarkable, with a 20-member commercialization team assembled within just two months. Prior to this, the company had only an R&D department. The establishment of the sales team facilitated product launches, and its management system aligns closely with that of mature multinational corporations. “Following the ‘4+7’ pilot program, the most significant change has been the ease of recruitment. People are beginning to recognize that domestic innovative pharmaceutical companies will soon emerge as a new force in the industry.”
Capital investment also surged rapidly. According to statistics from the VCBeat database, global financing in the healthcare sector reached RMB 157.1 billion in 2017, a year-on-year increase of 57%. At the end of 2017, significant adjustments were made to the listing rules of the Hong Kong Stock Exchange, which “allowed pre-revenue biotechnology companies to list in Hong Kong, with an expected minimum market capitalization of HKD 1.5 billion,” thereby opening a channel for public fundraising by new drug research and development enterprises.
Chen Haigang has clearly felt the market heating up. As the industry happens to be in a period of rapid growth, the valuations of his held projects have risen significantly compared to four years ago, leading to a substantial increase in the fund’s paper returns.
He even passed on certain projects due to high valuations. “R&D must adhere to objective scientific principles. While capital markets may currently assign lofty valuations, it is difficult to artificially accelerate the timeline for achieving specific clinical outcomes. Doing so risks overextending future potential… In the second half of 2018, we slowed our investment pace due to valuation concerns, causing us to miss out on some opportunities.”
Compared with PD-1, CAR-T therapy appears to have a more tumultuous fate.
The 2016 “Wei Zexi incident” sparked widespread skepticism toward cellular immunotherapy after the CIK therapy involved was proven ineffective, unfairly tarnishing the reputation of CAR-T technology, which had demonstrated promising clinical efficacy. This setback was further compounded when Juno Therapeutics abandoned its CAR-T product, JCAR015. Fortunately, in 2017, the FDA approved two CAR-T therapies—Novartis’s Kymriah and Kite/Gilead’s Yescarta—thereby restoring credibility to this novel treatment modality.
In early March, just before being interviewed by a reporter from 21CBR, Wang Liqun was discussing the “Two Sessions” with his management team; he expressed hope that delegates would offer recommendations on regulatory policies for cell therapy.
In late February, the National Health Commission drafted the Regulations on the Clinical Application Management of New Biomedical Technologies (Draft for Comments) and subsequently published on its official website the Reply to Proposal No. 4443 (Medical and Sports Category No. 434) of the First Session of the 13th National Committee of the Chinese People’s Political Consultative Conference. The reply stated that somatic cell therapy for refractory diseases such as tumors has demonstrated promising application prospects, and that “the National Health Commission will fully support the drug registration applications for cell therapy products, and organize the application, selection, and filing of clinical research institutions conducting cell therapy technologies.”
Fosun Kite is accelerating the commercialization of CAR-T therapy. In Wang Liqun’s view, the National Health Commission introduced the administrative measures to regulate clinical research on novel biomedical technologies, ensure medical quality and safety, and accelerate the clinical translation and application of leading-edge technologies. However, the draft for public comment fails to clearly define and differentiate “novel biomedical technologies,” particularly lacking classified management for somatic cell technologies.
He explained that cell therapies are categorized into high-risk and low-risk groups. As a high-risk product, CAR-T cell therapy must be submitted for approval and regulated as a pharmaceutical drug. Implementing a dual-track regulatory system for cell therapy products could lead to confusion regarding industry quality standards and clinical research norms, thereby hindering the standardized development of the industry. For patients, only CAR-T products under strict regulation can ensure their safety and interests.
“Previously, CAR-T companies had reached a consensus: dozens of Investigational New Drug (IND) applications had been submitted, and more than ten had been approved for clinical trials following review by the Center for Drug Evaluation (CDE) under the National Medical Products Administration. The CDE adopts a logic of ‘lenient entry but strict exit,’ allowing more cell therapy products to enter the pipeline with an open attitude, while rigorously verifying their eligibility for marketing authorization through robust clinical trials.” Wang Liqun believes that the existence of a dual-track system raises concerns. If the standards set by the National Health Commission are lower than those of the Center for Drug Evaluation, it could undermine the resolve of companies committed to developing high-quality cell therapy products through substantial investment. This would render the pharmaceutical regulatory framework recently established by the CDE ineffective, preventing promising cell therapies from being widely used in clinical practice to benefit patients, similar to other drugs. Such a scenario would deviate from the requirements of the International Council for Harmonisation of Technical Requirements for Pharmaceuticals for Human Use (ICH) and hinder alignment with international standards.
Another CAR-T industry practitioner expressed the same concern: “Under the current market access mechanism, many CAR-T products could reach the market within two to three years. Hastily relaxing approval criteria would do more harm than good.”
In contrast to Wang Liqun’s concerns about market access, Li Ning is closely monitoring the issue of rational drug use.
It is a common perception among Chinese patients that imported drugs are expensive but offer superior efficacy. To achieve domestic substitution, low price alone is insufficient; it is essential to establish a reputation for therapeutic efficacy, with clinical response rate serving as a key performance indicator. Truly achieving targeted therapy involves determining whether physicians can identify patients who meet the criteria for treatment, and whether those who stand to benefit can use the medication correctly.
Currently, Junshi Biosciences’ sales team comprises approximately 140 members. Li Ning’s directive is straightforward: to educate physicians on the drug’s risks, efficacy, and eligible patient criteria. “I aim for 100% of patients treated with Tuoyi to benefit from it. While stringent patient selection may impact short-term sales, it ultimately builds long-term brand equity.”
Li Ning told reporters that in the sales department of Junshi Biosciences, the focus of KPI assessment is on comparing therapeutic efficacy. “We track hospitals where academic promotion is conducted and monitor the effective rate after doctors prescribe medications to patients. For the same drug, if Sales Representative A achieves an effective rate of 50% while Sales Representative B only reaches 20%, this difference reflects whether the medication was appropriately indicated.”
Li Ning showed 21CBR reporters the internally used training materials, the "Blue Book" and the "Red Book," which specifically introduce the basic principles of tumor immunotherapy, treatment methods for different drugs, clinical manifestations, etc. "Tumor immunotherapy is a new thing, unlike targeted therapy and chemotherapy drugs that have been in use for over a decade; it has a shorter history of application, and even abroad, there is limited experience in its use. The industry has a responsibility to educate doctors and patients: first, it is not a miracle drug; second, it must be used correctly. The prospects are broad, but there is still a long way to go." Regarding the commercialization process, Li Ning was very cautious.
The concerns raised by Wang Liqun and Li Ning highlight the systemic challenges inherent in new drug development. The phrase “overtaking on a bend” has gained significant traction across various industries in China. When asked whether innovative pharmaceutical companies can achieve this, nearly all respondents offered a similar verdict: “The future is promising, but concerted efforts are needed to catch up.”
Li Ning believes that Western pharmaceutical companies are more advanced in pure biomedicine and basic research. “It is like having much more fertile soil, making it relatively easier to grow, blossom, and bear fruit. Our innovation itself suffers from considerable deficiencies at the foundational level.”
Furthermore, China is still playing catch-up in many areas of basic research and standard-setting. “Our technical review capabilities and the professionalism of our reviewers have improved rapidly, but we must not overlook the existing gaps.” Li Ning cited PD-1 inhibitors as an example: Tuoyi’s approval for market launch in China lagged behind Keytruda and Opdivo by only 3–6 months, which may seem like a short interval. In reality, extensive clinical studies had already been conducted for the latter two drugs, whereas China was just beginning its efforts. “This has led to issues of redundant research. If certain data have already become standardized, there is no necessity for latecomers to conduct additional clinical trials. However, such data standards remain currently absent.”
Liang Yi believes that the development trajectory of China’s pharmaceutical industry may follow a “three-step approach without a fixed timetable”: Step one is to excel in generic drugs; step two is to pursue innovation based on generics; and step three is to achieve dominance by innovative drugs. Throughout this process, success hinges on patience and focus—on staying committed to the right path and persevering. The alignment between capabilities and organizational structure determines the pace of development. “We are now at a stage where small streams converge into a river. It is essential to concentrate efforts on a single trend and avoid arbitrary pauses or shifts along the way. The straighter the path, the clearer the goal.”
“Always maintain a learning mindset; only when capabilities are fully developed across all areas will innovation naturally flourish.” Liang Yi stated candidly that amidst the commercial tide, there are many temptations, such as developing generic drugs with promising market prospects. “Our principle is to focus on innovative specialty drugs, which serves as both our foundation and the criterion for product selection.”
Junshi Biosciences is about to reap the rewards of its steadfast commitment.
“The market size for PD-1 inhibitors is substantial enough to accommodate a wide range of products. If a product possesses distinct characteristics and offers appropriate monotherapy and combination therapy options, there will be significant room for growth relative to the tens of millions of new and prevalent cancer patients,” said Li Ning. He believes that as more domestically produced PD-1 inhibitors enter the market with better cost-effectiveness, they could establish a 7:3 or even 8:2 market share ratio against imported drugs in the field of cancer treatment. “It is entirely possible for domestic drugs to dominate the market, provided that safety and efficacy are ensured.”
Wu Xiaobin stated that drug development carries substantial overall risk. “The current R&D landscape in China is highly favorable; it seems we have yet to hear of failures, or such cases are exceedingly rare. However, as our country continues to advance new drug development, we will inevitably face such situations sooner or later. At that time, will entrepreneurs and investors be psychologically prepared?”
