CDE Releases Draft Guidance on Using Real-World Evidence to Support Drug Development and Evaluation of Effectiveness and Safety
On May 29, VCBeat New Medicine (WeChat ID: biobeat1) learned that the Center for Drug Evaluation of the National Medical Products Administration had organized the drafting and release of the “Basic Considerations for Using Real-World Evidence to Support Drug Development (Draft for Comment).” To encourage the research and development of innovative drugs, and considering situations in which clinical trials are infeasible or difficult to conduct during the clinical development process, the use of real-world evidence to evaluate the efficacy and safety of drugs has emerged as a viable strategy and approach.
On April 4, 2019, the FDA approved a new indication for Pfizer’s Ibrance (palbociclib) based on real-world data (RWD): in combination with an aromatase inhibitor or fulvestrant, it is indicated for the treatment of male patients with HR-positive, HER2-negative metastatic breast cancer.
The FDA’s approval this time was based primarily on U.S. electronic health record data, as well as real-world medication data for male patients from the IQVIA insurance database, Flatiron Health’s breast cancer database, and Pfizer’s global safety database regarding post-marketing use of Ibrance, rather than on clinical trials or assessments of clinical performance. Typically, FDA drug approvals are based on open-label Phase III clinical trials; however, for drugs treating very rare conditions with notably significant efficacy, single-arm trials may serve as the basis for review. Even for biosimilar approvals, rigorous evaluation of equivalent clinical performance is required. This marks the first time the FDA has approved a drug indication based solely on real-world evidence.
Driven by the 21st Century Cures Act, the U.S. FDA successively issued documents such as “Use of Real-World Evidence to Support Regulatory Decision-Making for Medical Devices,” “Guidance on Using Electronic Health Record Data in Clinical Investigations,” and “A Framework for FDA’s Real-World Evidence Program” between 2017 and 2018.
In the drug development process, conventional clinical trials may be difficult to implement, entail prohibitive time costs, or raise ethical concerns in certain scenarios, such as rare diseases lacking effective treatments and life-threatening serious conditions. Consequently, in recent years, the use of real-world evidence (RWE) to evaluate the efficacy and safety of drugs has become an increasingly prominent focus in pharmaceutical research and development both domestically and internationally.
"Basic Considerations for Real-World Evidence Supporting Drug Development (Draft for Comment)" is positioned to support drug development. Based on the aforementioned background and existing experience, it puts forth fundamental considerations aimed at clarifying the relevant definitions of real-world studies in drug development, defining the status and scope of application of real-world evidence in drug development, and exploring evaluation principles for real-world evidence, with a view to providing scientific and feasible guidance for the industry in leveraging real-world evidence to support drug development.
The drafting group for this guideline was established under the tripartite academic coordination committee formed through the strategic partnership between the Center for Drug Evaluation (CDE) and Southern Medical University. Comprising representatives from academia, the pharmaceutical industry, and regulatory authorities, the group ensured the efficient and high-quality completion of this guideline. Officially launched in November 2018, the initiative included seminars held on January 12 and March 30, 2019. After six months of work, the draft for public comment was finalized, with the full content presented below:
Key Considerations for Real-World Evidence in Supporting Drug Development
I. Introduction
Randomized Controlled Trials (RCTs) are considered the “gold standard” for evaluating drug efficacy and are widely adopted in clinical drug trials. RCTs strictly control inclusion and exclusion criteria as well as other conditions, and employ randomization for group assignment. This approach minimizes factors that could affect causal inference, thereby yielding more definitive study conclusions and generating evidence with high reliability. However, RCTs have their limitations: stringent inclusion and exclusion criteria may reduce the representativeness of the trial population relative to the target population; the standardized interventions used may not fully align with real-world clinical practice; and limited sample sizes and short follow-up periods may lead to insufficient detection of rare adverse events. These limitations pose challenges when extrapolating RCT findings to actual clinical applications. Furthermore, for certain rare diseases and life-threatening serious conditions lacking effective treatments, conventional RCTs may be difficult to implement, require prohibitive time costs, or raise ethical concerns. Consequently, how to leverage Real World Evidence (RWE) in drug development, either independently or as supplementary evidence to RCTs, to evaluate drug efficacy and safety has become a issue of common concern and significant challenge for regulatory agencies, the pharmaceutical industry, and academia worldwide.
First, we need to conceptually clarify the definition and connotation of real-world evidence.
Secondly, whether and how Real World Data (RWD), which serves as the foundation for real-world evidence, can provide adequate support involves many issues that urgently need to be discussed, including data sources, data standards, data quality, data sharing, and data infrastructure.
Third, the regulatory vacuum. Currently, there are no mature international regulations in place. In the absence of established precedents, active exploration, research, and innovation are required to develop guidelines tailored to the realities of China’s pharmaceutical industry.
Fourth, the methodologies for evaluating real-world evidence require standardization. Real-world evidence is derived from the accurate and comprehensive analysis of real-world data, primarily employing causal inference methods. These methods involve complex models and model assumptions, corresponding covariate selection, identification of confounding factors, and definition of intermediate variables and instrumental variables. This places higher demands on statistical analysts and creates an urgent need for regulatory framework development.
Fifth, the scope of application for real-world evidence (RWE) needs to be clearly defined. The primary role of RWE is to complement evidence from traditional clinical trials, thereby forming a comprehensive and rigorous chain of evidence that enhances the scientific rigor and efficiency of drug development, rather than replacing such trials. Therefore, it is necessary to clearly define the scope of application for RWE based on the practical realities of drug development, with the flexibility to adjust this scope as circumstances evolve.
In view of the above reasons, this guideline aims to clarify the relevant definitions of real-world studies in drug development, specify the status and scope of application of real-world evidence in drug development, explore the evaluation principles of real-world evidence, and provide scientific and feasible guidance for the industry to utilize real-world evidence to support drug development.
The American Recovery and Reinvestment Act of February 2009 provided a significant boost to Comparative Effectiveness Research (CER; see Glossary). Against the backdrop of real-world settings inherent to CER, the concept of Real World Research/Study (RWR/RWS) was proposed.
In December 2016, the United States enacted the 21st Century Cures Act, which aims to encourage the U.S. Food and Drug Administration (FDA) to conduct research and utilize real-world evidence to support regulatory decision-making for drugs and other medical products, thereby accelerating their development. Driven by this legislation, the FDA successively issued the Guidance on Use of Real-World Evidence to Support Regulatory Decision-Making for Medical Devices, the Guidance on Using Electronic Health Record Data in Clinical Investigations, and the Framework for FDA’s Real-World Evidence Program between 2017 and 2018.
In 2013, the European Medicines Agency (EMA) issued the “Reflection Paper on Data-Driven Models of Disease Progression and Trial Evaluation in Alzheimer’s Disease,” which discussed the technical details of using real-world observational data to establish disease progression models. In 2014, the EMA launched the Adaptive Licensing Pilot project to assess the feasibility of using observational study data to support decision-making. In November 2016, it released the “Scientific Guideline on Post-Authorisation Efficacy Studies.”
The Pharmaceuticals and Medical Devices Agency (PMDA) of Japan proposed international harmonization on a new topic regarding technical requirements for more efficient use of real-world data in post-marketing pharmacoepidemiological studies at the level of the International Council for Harmonisation of Technical Requirements for Pharmaceuticals for Human Use (ICH).
China’s systematic efforts to leverage real-world evidence (RWE) in support of drug development and regulatory decision-making are still in their early stages. National drug regulatory authorities have already begun applying RWE in review practices; for instance, the 2018 approval of an expanded indication for bevacizumab in combination with platinum-based chemotherapy regimens relied on findings from three retrospective studies as RWE to support the final decision. Another example involves a post-marketing prospective, observational real-world study conducted for a certain drug to generate more robust evidence of its effectiveness and safety.
II. Definitions Related to Real-World Studies
Broadly defined, real-world studies encompass both research involving the general population and research involving clinical populations; the real-world evidence derived from the latter can be used to support medical product development and regulatory decision-making, as well as for other scientific purposes. This guideline is limited to real-world studies intended to support medical product development and regulatory decision-making (see figure below).
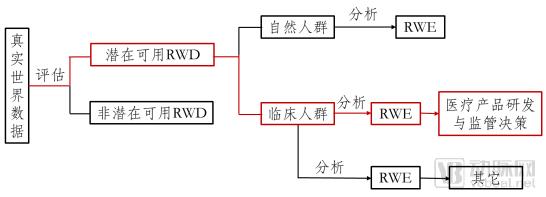
Figure 1. Pathway from Real-World Data (RWD) to Real-World Evidence (RWE) Supporting Regulatory Decision-Making for Medical Products
We define real-world studies as follows: collecting patient-related data in real-world settings (real-world data) and, through analysis, obtaining clinical evidence on the use value and potential benefits or risks of medical products (real-world evidence). The primary study type is observational research, although pragmatic clinical trials may also be conducted.
(1) Definition
Section 505F(b) of the U.S. Federal Food, Drug, and Cosmetic Act (FD&C Act) defines real-world data as “data relating to patient health status and/or the delivery of health care routinely collected from a variety of sources.” The FDA further elaborates on this definition in *Framework for Real-World Evidence Programs* and *Use of Real-World Evidence to Support Regulatory Decision-Making for Medical Devices*, characterizing real-world data as data derived from sources other than traditional clinical trials that pertain to drug utilization or potential benefits and risks. Examples include Electronic Health Record (EHR) data, Electronic Medical Record (EMR) data, medical claims data, data from product and disease registries, patient-reported data (including data from home settings), and data from other health monitoring devices (such as mobile devices).
We define real-world data as: data related to patients’ medication use and health status, and/or derived from various routine medical processes.
(2) Sources of Real-World Data
Common sources of real-world data in China include:
1) Health Information System (HIS): Similar to EMR/EHR, it comprises digital patient records containing both structured and unstructured data fields, such as patient demographics, clinical characteristics, diagnoses, treatments, laboratory tests, safety profiles, and clinical outcomes.
2) Health Insurance System: Contains data on structured fields such as patients’ basic information, healthcare service utilization, prescriptions, billing settlements, medical claims, and managed care.
3) Disease Registry System: A database of patients with specific diseases (typically chronic conditions), usually derived from hospital-based disease cohort registries.
4) China Adverse Drug Reaction Surveillance Sentinel Alliance (CASSA): Establishing an active surveillance and evaluation system for the safety of drugs and medical devices by leveraging electronic data from healthcare institutions.
5) Natural Population Cohort Database: Natural population cohort and disease-specific cohort databases that have been established or are currently being established in China can serve as potential real-world data.
6) Omics-related databases: Databases that collect omics-related information on patients’ physiology, biology, health, behavior, and potential environmental interactions, such as pharmacogenomics, metabolomics, and proteomics databases.
7) Death Registration Database: A database formed from death registrations jointly confirmed by hospitals, Centers for Disease Control and Prevention (CDC), and household registration authorities.
8) Data from mobile devices: Utilize mobile devices, such as wearable devices, to detect relevant data obtained from subjects.
9) Other special data sources: databases created for specific purposes, such as the National Immunization Program database.
(3) Data Quality Assessment
The quality of real-world data is primarily assessed through its relevance and reliability.
1) Relevance: Important relevant factors for assessing whether real-world data are suitable for regulatory purposes include, but are not limited to:
① Whether it includes important variables and information related to clinical outcomes, such as medication use, patient demographics and clinical characteristics, covariates, outcome variables, follow-up duration, and sample size;
② Whether the definitions of clinical outcomes are accurate and whether their corresponding clinical significance is clearly defined;
③ Whether the definition of the target population is accurate and representative;
④ Whether the study protocol and statistical analysis plan satisfy the validation of the study hypotheses.
2) Reliability: The reliability of real-world data is primarily evaluated in terms of data completeness, accuracy, quality assurance, and quality control.
① Completeness: While missing data is an inevitable issue in real-world data, the proportion of missingness should remain within certain limits. The extent of data missingness may vary across different studies. When the proportion of missing data for specific variables exceeds a predefined threshold, it introduces substantial uncertainty into the study conclusions. In such cases, careful consideration is required to determine whether the data are suitable for generating real-world evidence.
② Accuracy: The accuracy of data is of paramount importance and requires identification and verification against authoritative reference sources. For instance, blood pressure measurements must be performed using calibrated sphygmomanometers in strict adherence to operational protocols; additionally, endpoint events should be adjudicated by an independent Endpoint Adjudication Committee.
③ Quality Assurance: Quality assurance refers to measures for preventing, detecting, and correcting data errors or issues that arise during the research process. Closely related to regulatory compliance, quality assurance should permeate every aspect of data management. As a fundamental requirement, standard operating procedures (SOPs) must be established for each stage of data management.
④ Quality Control: Quality control must be implemented at every stage, including data collection, modification, transmission, storage, and archiving, as well as data processing, analysis, and submission, to ensure the accuracy and reliability of real-world data. To this end, it is necessary to establish a comprehensive, standardized, and reliable data management process or protocol.
(4) Data Standards
Data standards ensure that submitted materials are predictable and consistent, and are in a format usable by information technology systems or scientific tools. To enable real-world data from multiple sources to work together, the data must be converted into a common format with standardized representations (such as terminologies, glossaries, and coding schemes).
Furthermore, key factors to consider when assessing whether the quality of real-world data (RWD) is sufficient to support drug development include, but are not limited to: whether there are clear procedures and qualified personnel for data collection; whether a common definitional framework, i.e., a data dictionary, is used; whether a common timeframe for collecting key data points is adhered to; whether timelines have been established for research plans, protocols, and/or analysis plans related to RWD collection; whether the technical methods for capturing data elements are adequate, including integration of data from various sources, recording of medication use data, and linkage with claims data; whether patient selection minimizes bias to accurately represent the true target population; whether data entry and transmission ensure availability and timeliness; and whether adequate and necessary patient protection measures are in place, such as safeguarding patient privacy and obtaining informed consent in compliance with regulatory requirements.
Real-world evidence (RWE) is clinical evidence regarding the usage and potential benefits or risks of medical products, derived from the analysis of real-world data. This definition is not conceptually limited to evidence obtained through retrospective observational studies; it also allows for the prospective collection of broader data to generate evidence, particularly including study designs such as Pragmatic Clinical Trials (PCTs).
III. Scenarios in Which Real-World Evidence Supports Drug Development and Regulatory Decision-Making
Real-world evidence can support drug development in various forms, covering multiple stages including pre-approval clinical development and post-marketing re-evaluation. Any use of real-world evidence for the purpose of drug registration requires prior and sufficient communication with regulatory authorities to ensure mutual alignment on study objectives and methodology.
In clinical trials for rare disease therapies, beyond the challenges of scarce cases and difficult recruitment, the greatest hurdle lies in the selection of controls, as there are often no or few available treatment options for rare diseases. Therefore, real-world data derived from natural history cohorts can serve as external controls.
External controls are primarily used in non-randomized single-arm trials and can be either historical or concurrent. Historical external controls utilize real-world data obtained previously as the comparator, whereas concurrent external controls use disease registry data collected during the same period as the single-arm trial. When employing external controls, careful consideration must be given to the impact of heterogeneity and comparability of the target population on the validity of real-world evidence.
For drugs already on the market, long-term clinical practice may reveal the need to expand their indications. Typically, randomized controlled trials (RCTs) are used to support such expansions. However, when RCTs are not feasible or fail to generate optimal evidence, pragmatic clinical trials (PCTs) represent a viable alternative. For instance, clinical practice may suggest that a new drug developed for diabetes could offer potential benefits to patients with cardiovascular diseases, such as heart failure. In such cases, employing an RCT design would make participant recruitment extremely difficult and could even raise ethical concerns, whereas a PCT design may prove more feasible.
In the field of pediatric medication, off-label drug use is common in clinical practice in China. Leveraging real-world evidence to support the expansion of indicated populations is also a strategy for drug development.
A typical case of using real-world evidence (RWE) to support the expansion of combination therapy indications is bevacizumab, a humanized monoclonal antibody targeting vascular endothelial growth factor (VEGF). In 2015, bevacizumab was approved in China for first-line treatment of patients with unresectable advanced, metastatic, or recurrent non-squamous non-small cell lung cancer (NSCLC) in combination with chemotherapy (carboplatin and paclitaxel). However, in real-world clinical practice, the chemotherapy regimens combined with bevacizumab were not limited to carboplatin and paclitaxel, but also included pemetrexed plus platinum agents, gemcitabine plus cisplatin, and others. In October 2018, the approval for bevacizumab was expanded to include combination with platinum-based chemotherapy regimens, strongly supported by findings from three real-world studies. These studies retrospectively analyzed patient data from three hospitals, all demonstrating that adding bevacizumab to platinum-based doublet chemotherapy significantly prolonged progression-free survival (PFS) and overall survival (OS) compared with chemotherapy alone. The results were consistent with global population data, and no new safety concerns were identified. Furthermore, relevant real-world studies provided efficacy data across different patient subgroups, such as those with EGFR mutations and brain metastases, thereby confirming the effectiveness and safety of bevacizumab-based combination therapy from multiple perspectives.
Drugs approved based on randomized controlled trial (RCT) evidence often suffer from limitations such as limited safety information, uncertainty in generalizing efficacy conclusions, suboptimal dosing regimens, and a lack of pharmacoeconomic data. These shortcomings arise due to small sample sizes, short study durations, strict inclusion criteria for participants, and standardized interventions. Therefore, it is necessary to leverage real-world data to conduct more comprehensive evaluations of the drug’s effectiveness, safety, dosing regimens, and economic benefits in natural populations, and to continuously adjust decision-making based on real-world evidence.
For example, a certain drug is a cardiovascular medication that has already been approved in more than 50 countries and regions worldwide. In the international multicenter clinical trials conducted for this drug, the efficacy results in the Chinese population showed high variability due to the small sample size of the Chinese subgroup, the limited number of cardiovascular events, and the short duration of drug exposure. As a drug already marketed overseas and urgently needed for clinical use, to further clarify its efficacy in Chinese patients, the applicant plans to conduct a prospective, observational, real-world study after marketing approval. This study aims to evaluate the preventive effect of the drug combined with standard therapy versus standard therapy alone on major cardiovascular events in Chinese patients with cardiovascular disease.
It is a unique phenomenon in China that traditional Chinese medicine (TCM) hospital preparations have been widely used in clinical practice for extended periods without obtaining marketing approval. For the clinical development of such drugs, integrating real-world studies with randomized controlled trials will help establish scientifically sound and feasible pathways for clinical development and provide evidence to support regulatory decision-making for TCM hospital preparations.
There are various strategies for leveraging real-world evidence (RWE) to support the research and development of hospital preparations of traditional Chinese medicine (TCM). Figures 2 and 3 illustrate two potential pathways. The pathway combining observational studies with randomized controlled trials (RCTs) is shown in Figure 2. In the first phase, a retrospective observational study is conducted. During this phase, historical real-world data related to the use of the drug should be collected as comprehensively as possible, including all potential covariates; data cleaning rules should be established; potential controls should be selected; data quality should be assessed; and appropriate statistical methods should be employed for comprehensive and detailed analysis. If the retrospective observational study indicates that the drug offers potential benefits to patients in clinical practice, the study may proceed to the next phase; otherwise, it is terminated. The second phase involves a prospective observational study. Building on the foundation laid in the first phase, the design of the prospective observational study can be more rigorous, encompassing data collection systems, data quality control, data cleaning rules, and clearly defined controls. At a certain stage of the prospective observational study, if the data analysis results are consistent with those of the retrospective observational study and continue to demonstrate significant clinical benefits for patients, a third-phase RCT may be initiated in parallel. The RCT may begin as an exploratory trial; however, if the evidence from prior observational studies is sufficiently robust, a confirmatory RCT may be conducted directly. From a temporal perspective, the duration of the RCT is encompassed within the timeline of the prospective observational study. The latter may conclude simultaneously with the RCT or extend for a period thereafter, depending on whether the real-world evidence is deemed sufficient.
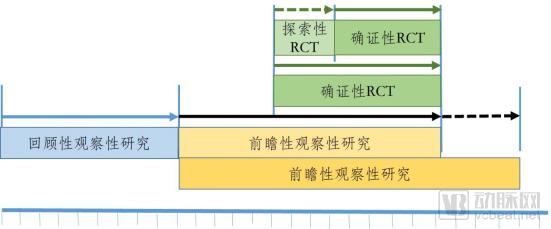
Figure 2. One of the Path Strategies for the Research and Development of Hospital Preparations of Traditional Chinese Medicine
Figure 3 illustrates the integrated approach combining observational studies with Pragmatic Clinical Trials (PCTs). In the first phase, a retrospective observational study is conducted. If the results indicate potential clinical benefits for patients, the research proceeds to the next phase; otherwise, the study is terminated. The second phase involves conducting a PCT, the evidence from which can be used to support the evaluation of clinical efficacy and safety.
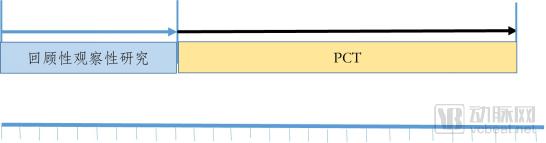
Figure 3. The Second Path Strategy for the R&D of Hospital Preparations of Traditional Chinese Medicine
Using real-world evidence to guide clinical study design has more practical applications. For instance, the two development pathways for hospital preparations of traditional Chinese medicine cited in the previous section both leveraged real-world evidence generated from retrospective observational studies. This evidence included data on the natural history of the disease, its prevalence in the target population, the efficacy and effectiveness of standardized treatments, and the distribution and variation of key covariates associated with efficacy and effectiveness within the target population, thereby providing a basis for designing subsequent studies. More broadly, real-world evidence can serve as an effective reference for defining inclusion and exclusion criteria, estimating parameters for sample size calculations, and determining non-inferiority margins.
Precision medicine aims to better predict the therapeutic benefits and risks of drugs for specific populations (subgroups). Real-world evidence derived from real-world data makes this possible. For instance, due to limited sample sizes, traditional clinical trials often overlook or fail to adequately address subgroup effects in their study protocols. This results in a failure to fully capture critical information about potential treatment responders or high-risk populations prone to severe adverse effects, thereby leading to inaccurate targeting of the intended population. Through comprehensive analysis of real-world data, it is possible to thoroughly evaluate the therapeutic benefits and risks across different subgroups, thereby generating real-world evidence to support more precise target population identification.
In preclinical and early-stage clinical studies of targeted therapies, biomarker identification is critical. By leveraging real-world data—including omics data from population cohorts, public genomic databases, and relevant clinical records—and applying various machine learning-based target-oriented analytical techniques to generate real-world evidence, precise patient population targeting for targeted therapeutic agents can be supported.
IV. Basic Design of Real-World Studies
Pragmatic Clinical Trial (PCT), also known as practical clinical trial, refers to a clinical trial that closely mimics real-world clinical settings. It is a study design situated between randomized controlled trials (RCTs) and observational studies. Unlike RCTs, PCTs may employ either standardized or non-standardized interventions; participants may be assigned through randomization or natural selection; inclusion criteria are broader, enhancing the representativeness of the target population; and the evaluation of intervention outcomes extends beyond clinical efficacy and safety. In contrast to observational studies, PCTs are interventional in nature, albeit with considerable flexibility in intervention design.
Because pragmatic clinical trials (PCTs) must account for the influence of all potential factors, including various biases and confounders, their study design and statistical analysis are relatively complex, and the required sample size typically far exceeds that of randomized controlled trial (RCT) designs. If PCTs employ randomization, they can reduce the impact of confounding and other biases, thereby providing robust causal inference. Furthermore, PCTs generally do not use blinding in most cases; therefore, sufficient attention must be paid to how to estimate and correct for the resulting detection bias. As studies conducted in settings closer to real-world clinical practice, the evidence generated by PCTs is regarded as the best and most feasible form of real-world evidence compared with other study types.
The use of external controls has limitations, primarily including differences in healthcare settings, changes in medical technology over time, varying diagnostic criteria, differing outcome measures, variations in patient baseline characteristics, diverse interventions, and difficulties in ensuring data quality. These limitations pose challenges to the comparability of study subjects, the precision of study results, and the reliability and generalizability of study conclusions.
To overcome or mitigate these limitations, it is essential first to ensure that the collected data meet the relevant quality standards for real-world data. Second, in terms of study design, a parallel external control design is preferable to a historical control design; prospective parallel external controls can adopt a disease registry model to ensure that data recording is as complete and accurate as possible. Third, appropriate statistical analysis methods should be employed, such as the rational use of Propensity Scores (PS) and Virtual Matched Control methods.
Data collected from observational studies are undoubtedly closest to real-world conditions; however, their primary limitations include various biases, difficulties in ensuring data quality, and challenges in identifying both observed and unobserved confounding factors, which introduce substantial uncertainty into the study conclusions.
Whether the data collected in observational studies are suitable for generating real-world evidence to support regulatory decisions, key considerations include: ① What are the data characteristics? (e.g., data collection for relevant endpoints, consistency of recording, and description of missing data) ② What are the features of the study design and analysis? (e.g., Is there an appropriate active control? Given potential unmeasured confounding factors and potential measurement variability, is a non-inferiority design applicable?) ③ Which sensitivity analyses and statistical diagnostic methods have been prespecified for analyzing real-world data?
Causal inference is the key technique for analyzing real-world data in observational studies. Commonly used statistical analysis methods in real-world studies are provided in Appendix 2.
V. Evaluation of Real-World Evidence
The evaluation of real-world evidence should adhere to two primary principles: whether the real-world evidence can support the scientific questions that need to be answered, and whether the required real-world evidence can be derived from existing real-world data through scientific analysis.
1. Real-World Evidence and the Scientific Questions It Supports
Before deciding to use any evidence, including real-world evidence (RWE), the scientific question that needs to be answered should first be clearly defined. Examples include safety considerations for the combined use of a drug with other medications after market approval; studies on expanded indications for approved products; and establishing robust and reliable historical controls for single-arm clinical trials. In this context, the original intention behind using RWE should be considered: is it because the corresponding scientific question pertains to the real-world setting, or is it because, although the question is oriented toward clinical research, traditional clinical trials cannot be effectively conducted? If it is the latter, whether RWE can replace traditional clinical trials to answer the same questions and yield robust conclusions will serve as an important criterion for evaluating the application of RWE.
2. How to Generate Real-World Evidence from Real-World Data
Answers to this question should generally include at least the following key features: ① research settings and data collection that approximate real-world conditions, such as more representative target populations, diverse interventions aligned with clinical practice, and natural selection of interventions; ② appropriate controls; ③ more comprehensive outcome evaluation; ④ effective bias control, such as the use of randomization and standardized measurement and assessment methods; ⑤ appropriate statistical analysis, including the correct application of causal inference methods, reasonable handling of missing data, and adequate sensitivity analyses; ⑥ reasonable interpretation of results; and ⑦ consensus among all stakeholders.
Finally, it is particularly important to emphasize that all study designs, hypotheses, and specific definitions related to the generation of real-world evidence should be clearly articulated in the study protocol in advance. Meanwhile, any use of real-world data and evidence with the ultimate goal of drug registration requires sufficient communication with regulatory authorities beforehand to ensure mutual consensus on research objectives, methodologies, and other aspects. Post-hoc remediation involving data citation, definitions, analysis, and interpretation is generally not accepted for regulatory decision-making.