What Makes a Me-Too Strategy Attractive to Investors? Insights from Atorvastatin and Beyond
Author: Chen Feng, Youxuan Capital
Reposted from BiotechVentureCapital
Recently, in discussions with industry peers, I have observed a phenomenon: many investors in new drugs are keen on “staking out territories.” For instance, they feel compelled to invest in at least one company in the PROTAC space, establish a foothold in oncolytic viruses, place a strategic bet in gene therapy, and similarly cover bispecific antibodies, cell therapies, and other areas. After extensive exchanges, I found their reasoning largely unconvincing. The predominant consensus is simply that if a trend is visible, one must position themselves accordingly. Frankly, I am not fond of this approach to decision-making.
No matter how impressive the technology platform, what good is it if it cannot be developed into a drug?
Drugs are meant to treat diseases. Regardless of the technology involved—even if a drug candidate is discovered by sheer luck—if it ultimately becomes an approved medication that addresses unmet clinical needs, it is a good drug.
I am not sure whether the term “track” originates from the internet or TMT sector, but it seems that domestic investors have grown accustomed to using this concept to frame and evaluate all issues. Can we interpret it this way: when a VC investor describes their investment in a particular track as “strategic positioning,” does it imply that they have not truly understood what they are investing in?
Nowadays, the “Me-too” strategy is often met with disdain and dismissal. However, many investors are hesitant to back First-in-class assets due to underlying uncertainties. This uncertainty does not stem from doubts about whether a drug can ultimately be developed, but rather from the fear of becoming a subject of ridicule in casual conversations. Consequently, many investors flock to Fast-follow projects in a herd-like manner. In my view, the Me-too strategy is not entirely unviable; it requires careful and in-depth analysis rather than blind conformity.
Before summarizing what constitutes a sound “me-too” strategy, let us first revisit the well-worn example of atorvastatin (Lipitor); a careful examination of its preclinical and clinical details offers valuable insights. (Here, “me-too” is defined as drugs targeting the same mechanism and indicated for the same condition as an already marketed product.)
The prevailing consensus is that the market for a given indication is limited, and the industry generally believes that the first three drugs targeting the same mechanism to reach the market have a higher probability of profitability. In this light, the success of atorvastatin, which ranked fifth, indeed appears somewhat anomalous.
Detailed Analysis of the Reasons for Success:
Primary Objective: Significantly Enhance Drug Potency. The development of atorvastatin faced a critical decision: whether to develop a single enantiomer or a racemate. Researchers spent over two years addressing numerous challenges in chiral synthesis and manufacturing processes. However, this investment proved worthwhile, yielding significantly greater efficacy. Had they settled for the quicker market entry of the racemate, the drug might have demonstrated only mediocre performance, similar to fluvastatin.
In the Phase I single-ascending-dose clinical trial of atorvastatin, healthy subjects tolerated doses up to 80 mg well. Only one subject in the 120 mg dose group experienced mild, transient agitation, excitement, and confusion, which were considered dose-limiting adverse effects. The mean plasma concentration, peak plasma concentration (Cmax), and area under the curve (AUC) demonstrated strong dose-dependency. The plasma half-life exceeded 14 hours, significantly longer than that of other statins. Atorvastatin is primarily metabolized in the liver, and its two major primary metabolites exhibit inhibitory activity comparable to that of the parent compound. Consequently, the inhibition of HMG-CoA reductase by atorvastatin in humans can be maintained for 20–30 hours.
In the multiple-dose escalation trial, doses ranging from 0.5 to 80 mg/day were administered once or twice daily for 14 consecutive days. The drug was well tolerated by subjects. Measurements revealed dose-dependent reductions in total plasma cholesterol (TPC) and low-density lipoprotein cholesterol (LDL-C) levels. Compared with the placebo control group, LDL-C levels decreased by 22.4%, 30.5%, 39.2%, 46.7%, and 57.8% in the 2.5 mg, 10 mg, 20 mg, 40 mg, and 80 mg groups, respectively, with highly statistically significant differences (p < 0.0013).
The results exceeded expectations. At the time, Black, the company’s Vice President of Clinical Research, believed that in terms of lowering LDL-C, the 10 mg dose had already demonstrated efficacy comparable to the highest FDA-recommended doses (40 mg) of other statins, while the 80 mg dose was 40% more potent than the highest doses of other statins.
Black therefore proposed an “all-or-nothing” strategy: designing the Phase II and III clinical protocols with a starting dose of 10 mg to demonstrate that the efficacy of this low dose was comparable to or better than high doses of other statins, and setting 80 mg as the maximum dose to showcase superior efficacy, thereby seeking FDA approval for the 80 mg dose in patients with severe hypercholesterolemia. Although this approach carried the risk of unpredictable toxic side effects from the high dose in a broader patient population, if successful, it would ensure that their product decisively outperformed other statins. This was critically important for Warner-Lambert, which was facing a crisis of declining growth.
Familial hypercholesterolemia is the most common and severe monogenic disorder of lipid metabolism, with an incidence of homozygous FH of approximately 1 in 1,000,000. Patients have LDL cholesterol levels that are on average three times higher than those of healthy individuals; most develop symptoms of coronary heart disease around the age of 10 and die from cardiovascular disease before the age of 30.
To secure marketing approval as quickly as possible and obtain a priority review voucher, Parke-Davis decided to evaluate atorvastatin for the treatment of patients with homozygous familial hypercholesterolemia (HoFH). Previously, Merck had engaged Dr. Frederick Raal in Johannesburg, South Africa, to assess the efficacy of simvastatin for this condition, but the study found no significant reduction in LDL cholesterol. In contrast, Parke-Davis’s small-scale trial of atorvastatin proved highly successful: participants experienced a 17% reduction in LDL cholesterol at a 40 mg dose and a 28% reduction at an 80 mg dose (p < 0.01). During the trial, Dr. Raal also demonstrated inhibition of cholesterol synthesis by measuring urinary mevalonic acid (MVA) accumulation. This marked the first time Dr. Raal had identified an effective pharmacological therapy for HoFH, and these findings contributed to the FDA granting atorvastatin a priority review voucher.
In its clinical trial dose-ranging design, atorvastatin took the risk of proposing and implementing an 80 mg “decisive” regimen, supported by a pivotal head-to-head CURVES clinical study.
This multicenter, randomized, open-label, parallel-controlled trial lasted for 8 weeks and compared the efficacy of atorvastatin at doses of 10 mg, 20 mg, 40 mg, and 80 mg with simvastatin (10 mg, 20 mg, 40 mg), pravastatin (10 mg, 20 mg, 40 mg), lovastatin (20 mg, 40 mg, 80 mg), and fluvastatin (20 mg, 40 mg) at their respective doses. A total of 534 patients with hypercholesterolemia aged 20–80 years were enrolled. The primary efficacy endpoint was the change in LDL cholesterol levels from baseline to week 8 of treatment. Changes in total cholesterol (TC), triglycerides (TG), and HDL cholesterol were also compared. The results are shown in the table below (note: “Avastatin” in the table refers to atorvastatin).
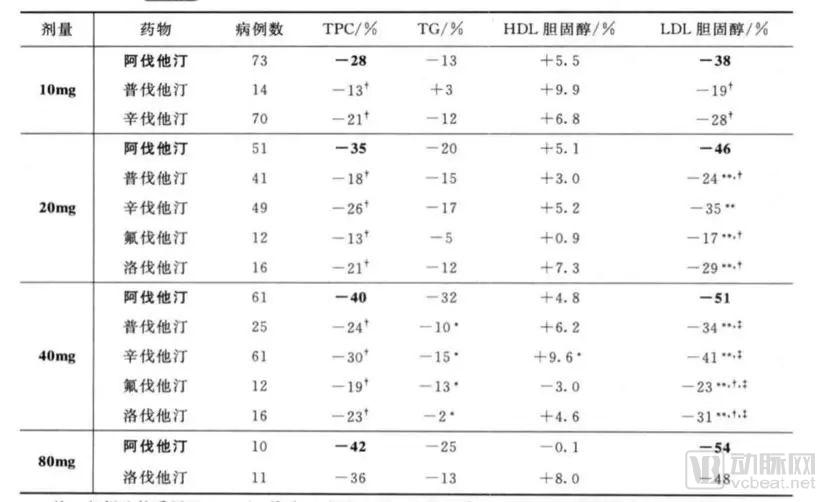
Atorvastatin at doses of 10 mg, 20 mg, and 40 mg reduced LDL cholesterol by 38%, 46%, and 51%, respectively, which were all greater than the reductions achieved with corresponding doses of simvastatin, pravastatin, lovastatin, and fluvastatin (P < 0.01).
Atorvastatin 10 mg produces efficacy equivalent to or greater than that of simvastatin 10 mg, 20 mg, and 40 mg; pravastatin 10 mg, 20 mg, and 40 mg; lovastatin 20 mg and 40 mg; and fluvastatin 20 mg and 40 mg (p < 0.01).
The clinical protocol of the CURVES trial was flawless, and the results were nearly perfect as well. With a priority review voucher, the FDA approved atorvastatin for marketing in the United States that December, including oral tablets in four strengths: 10 mg, 20 mg, 40 mg, and 80 mg.
Based on this, I believe a good Me Too strategy should possess the following characteristics:
Currently, many "me-too" drugs show little difference from already marketed medications, at best demonstrating non-inferiority. Since these follow-on products have only recently been launched, their real-world clinical issues have not yet fully emerged.
Taking ibrutinib as an example, it seems that everyone assumes that, compared with acalabrutinib, ibrutinib has improved selectivity and resolved some clinical side effect issues. Is it really that simple?
In terms of efficacy, Professor Lu Hua Wang from the University of Texas MD Anderson Cancer Center, a global leader in research on multiple BTK inhibitors such as ibrutinib and acalabrutinib, has previously commented.
The overall response rate (ORR) and complete response (CR) rate in the international, multicenter clinical trial of ibrutinib for mantle cell lymphoma (MCL) were 68% and 20%, respectively. In contrast, the international, multicenter clinical study of acalabrutinib for MCL reported an ORR of 80% and a CR rate of 40%. However, this does not necessarily indicate that acalabrutinib is more efficacious than ibrutinib. In the ibrutinib clinical trial (PCYC-1104), patients had received a median of three prior lines of therapy, meaning they were undergoing fourth-line treatment on average when starting ibrutinib. By comparison, patients enrolled in the acalabrutinib study had received a median of two prior lines of therapy, which naturally accounts for the higher ORR and CR rates observed. Subgroup analysis of the ibrutinib clinical study showed that among MCL patients who had received only one prior line of therapy, the ORR was 82%, comparable to that seen with acalabrutinib.
Therefore, in the absence of randomized controlled trials, it is not possible to determine which drug has superior efficacy.
Furthermore, the criteria for evaluating complete response (CR) efficacy differ between studies of ibrutinib and acalabrutinib. Ibrutinib studies utilized the 2007 Cheson IWG response criteria, whereas acalabrutinib studies adopted the 2014 Lugano classification system, which tends to yield higher CR rates.
Professor Wang Luhua also participated in the global clinical trials of BeiGene’s zanubrutinib. The patient enrollment profile was more similar to that of acalabrutinib, with the majority of enrolled patients receiving second-line therapy, although some were treated in the first-line setting. The efficacy evaluation system adopted in the trial was the 2014 Lugano classification, showing an overall response rate of approximately 80%, which is comparable to acalabrutinib. Due to differences in baseline characteristics of enrolled patients and inconsistencies in efficacy evaluation systems, there is currently no clinical evidence to demonstrate a difference in efficacy among these agents. Based on Professor Wang Luhua’s clinical experience, the three drugs exhibit similar clinical efficacy.
Both acalabrutinib and zanubrutinib have initiated head-to-head trials against ibrutinib, engaging in fierce competition. Without robust and credible comparative clinical data, physicians will not readily alter their prescribing practices.
Regarding side effects: headache is specific to acalabrutinib. The toxicities and adverse reactions of different BTK inhibitors vary, allowing clinicians to differentiate among them. For patients with severe headaches, acalabrutinib should be avoided in favor of ibrutinib; conversely, for patients with prominent rash, acalabrutinib may be recommended.
On Off-Target and Selectivity Issues
In fact, there is widespread discussion about whether the disadvantages of ibrutinib are due to increased side effects caused by off-target inhibition (of EGFR, JAK, and TEC). Could it be that its multi-target profile is precisely what leads to superior efficacy? Are these truly off-target effects or beneficial multi-target actions? Let us carefully examine whether these so-called off-targets have any value in the treatment of lymphoma.
Dysregulation of the EGFR/PI3K/Akt signaling pathway is closely associated with tumorigenesis and progression, and represents one of the important therapeutic targets in lymphoma. Aberrant activation of the JAK/STAT signaling pathway plays a critical role in the pathogenesis and development of various types of lymphoma.
In 2016, *Blood* revealed the distinct mechanisms underlying different subtypes of diffuse large B-cell lymphoma (DLBCL). As shown in the figure below, pure BTK inhibitors are effective only against the activated B-cell-like (ABC) subtype of DLBCL, while they have no mechanistic effect on the germinal center B-cell-like (GCB) subtype. However, ibrutinib has demonstrated efficacy in a small subset of GCB-DLBCL cases, which may be attributable to its multi-target effects.
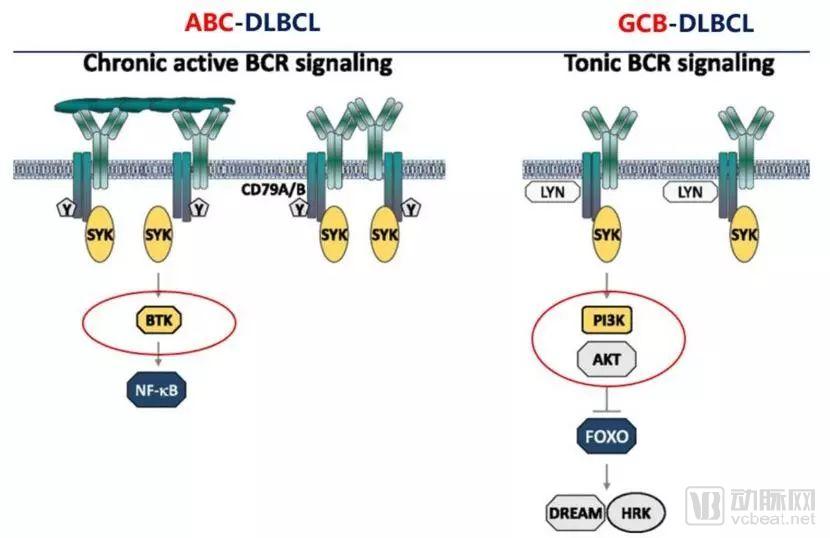
Therefore, EGFR, JAK/STAT, and TEC all play important roles in the development and progression of lymphoma. A variety of drugs targeting their upstream and downstream pathways are currently undergoing clinical trials, such as Idelalisib and AG490. Theoretically, targeting these pathways can lead to more effective treatment of lymphoma. Ibrutinib exhibits multi-target properties; in addition to inhibiting BTK, it also acts on certain upstream targets of PI3K. Its efficacy in a subset of patients with GCB-DLBCL further demonstrates the therapeutic potential of this multi-target approach.
For other BTK inhibitors with more single-target specificity, the likelihood of developing resistance during long-term treatment may be higher. Since currently available data are all short-term, longer follow-up data are needed to validate their efficacy in indolent lymphoma.
On the Issue of Drug Resistance
Irreversible covalent binding to the C481S site, as seen with acalabrutinib and zanubrutinib, is no longer a viable expectation. Current data suggest that ARQ531 holds the greatest promise. Notably, ARQ531 has garnered significant interest due to its efficacy against both wild-type BTK and the C481S mutation. Drawing a parallel to the trajectory of osimertinib, one can anticipate ARQ531’s future prominence in the field. We look forward to forthcoming clinical data.
In summary, for marketed oncology drugs such as ibrutinib, the clinical issues that have emerged to date—whether off-target side effects, enhanced efficacy through high selectivity, or drug resistance—are not significant. Therefore, developing “me-too” drugs following such blockbuster products is not a wise strategy for small biotechnology companies.
The primary reason is that the clinical endpoints for oncology drugs are either overall survival (OS) or progression-free survival (PFS), making it difficult to make clear predictions during Phase I and Phase II clinical trials, while objective response rate (ORR) shows no correlation with OS. If we could encounter a scenario similar to atorvastatin, where drug efficacy can be clearly assessed in Phase I clinical trials based on various biomarker indicators in healthy individuals, it would greatly facilitate the assessment of success probabilities for both drug developers and investors. This would avoid the half-guessing, half-reasoning approach fraught with anxiety that characterizes oncology drug development.
Tumor heterogeneity, tumor evolution, and mechanisms of drug resistance remain under continuous investigation. Faced with a disease adversary that is constantly changing and evolving, it is difficult for "Me-Too" strategies to achieve significant differentiation.
Moreover, competition in oncology therapeutics is extremely fierce. Candidate products with diverse targets, mechanisms, and modalities, along with a constant stream of novel therapies, are all vying for a share of the market, making it exceptionally difficult to emerge victorious. In pursuing differentiated strategies to overcome resistance for “me-too” drugs, the targeted proteins that have been cultivated for years could face a severe setback if PROTAC technology achieves success at any point.
This point is also crucial. Some “me-too” candidate products compete on selectivity, while others differentiate themselves through structural differences in their compounds; any distinct features will be prominently highlighted. Even if there is no pharmacological differentiation, efforts will still be made to identify distinctions at the level of compound structure.
Setting aside these various distracting factors, we must focus on the primary contradiction. If the clinical issue stems from an insufficiently wide therapeutic window, then this aspect warrants in-depth exploration. If the clinical issue is caused by other factors, it requires case-by-case analysis. The therapeutic window is the key to everything; thus, during Phase I clinical trials, we will have sufficient data to support and inform our judgments.
Reference:
——[ASH On-Site Update] Professor Wang Luhua Provides In-Depth Commentary on the Efficacy and Safety of Different BTK Inhibitors, Oncology Information
—— Case Studies in New Drug Development by Xia Guangxin and Shen Jingkang
——“Off-Target or Multi-Target? BTK Inhibitors Stir Up Controversy Again” WeChat Official Account: Xiao Dong Reads MM