Pfizer-Spun Rare Disease Biotech SpringWorks Therapeutics Launches U.S. IPO

SpringWorks Therapeutics
Disease Treatment Drug Developer
On September 13, 2019 (U.S. time), SpringWorks Therapeutics, a rare disease drug development company, listed on the Nasdaq. The IPO price was $18.00 per share, with an opening price of $24.50 and a real-time trading price of $22.63, representing a surge of 45.22%. This “new company,” spun off from Pfizer in 2017, completed its initial public offering just two years after its establishment, indicating that the secondary market has responded quite positively to the niche segment of rare diseases.
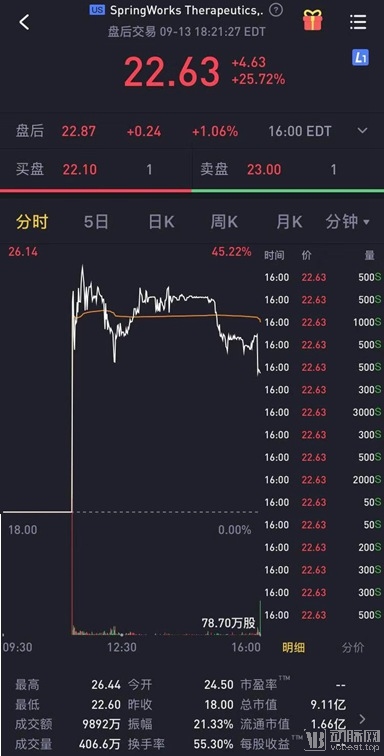
SpringWorks’ stock closed at $22.63 on its trading debut, with its market capitalization approaching $1 billion.
Since the FDA enacted the Orphan Drug Act in the 1980s, rare disease drug development has remained a priority for regulatory agencies worldwide. Policy incentives and untapped market opportunities have served as a beacon for companies navigating the challenging path of orphan drug R&D. Following policy-driven momentum, this sector appears to have identified new directions for growth, with SpringWorks Therapeutics emerging as a key player.
SpringWorks Therapeutics is a clinical-stage biopharmaceutical company focused on the development of therapies for rare diseases and oncology. Spun off from Pfizer in October 2017, it primarily assumed responsibility for the development of certain rare disease drug candidates from Pfizer’s portfolio. Divesting business lines that are less aligned with corporate strategy into independent entities has become a common practice among pharmaceutical giants in recent years; however, this does not imply that these assets lack investment value. Judging by SpringWorks’ fundraising track record to date, both pharmaceutical companies and investors remain highly interested in its drug pipeline, with Pfizer itself continuing to provide capital infusion to SpringWorks.
SpringWorks’ investors include leading global pharmaceutical companies such as Pfizer and GlaxoSmithKline (GSK), as well as prominent investment firms like Bain Capital and OrbiMed. Upon its establishment in September 2017, the company completed a $103 million Series A financing round, followed by a $125 million Series B round in April 2019. According to its prospectus, SpringWorks issued 7,352,941 ordinary shares at an offering price of $18 per share in this IPO, raising a total of $132 million. The proceeds will be used to support the clinical development of its drug candidates, nirogacestat and mirdametinib.
SpringWorks Revenue Table
SpringWorks Cash Flow
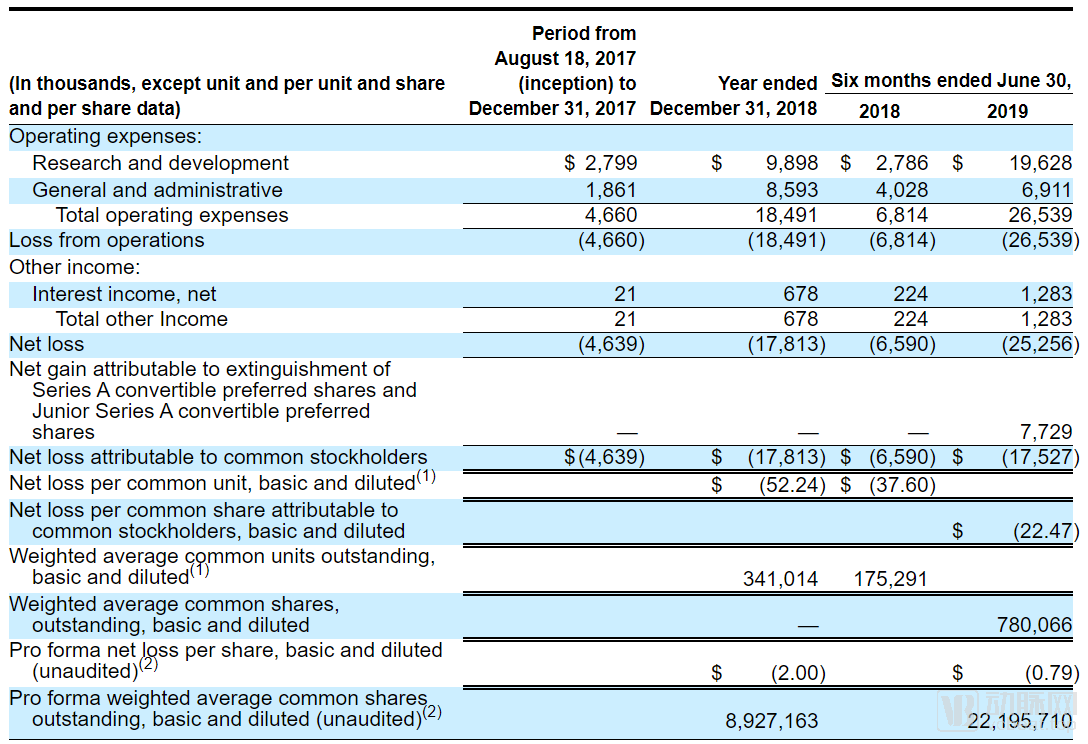
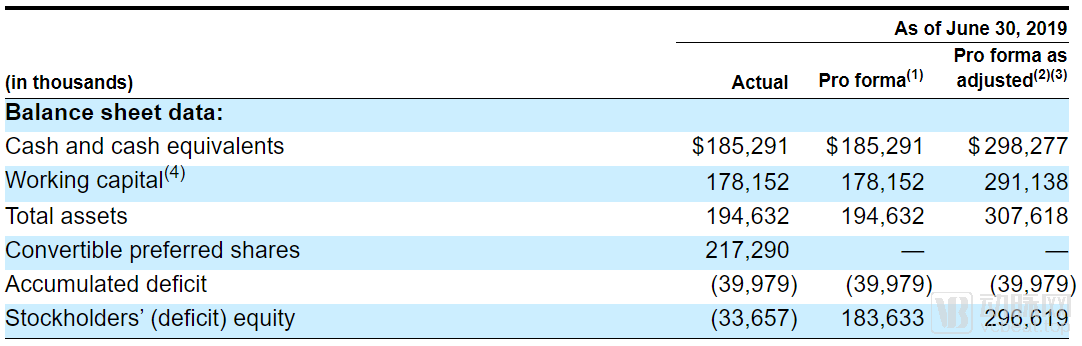
For start-up pharmaceutical companies, operating at a loss is the norm. SpringWorks Therapeutics reported a net loss of $17.8 million in 2018, but this appears to be just the beginning of its cash burn. With the launch of two new clinical trials, R&D spending surged in 2019, reaching $25.3 million in the first half alone—exceeding the full-year net loss for 2018. However, investors need not worry for now: SpringWorks still holds $185 million in cash on its balance sheet, and following this IPO, its cash reserves are expected to surpass $300 million. This should provide sufficient runway for SpringWorks to sustain operations until its products reach the market.
The product pipelines of rare disease drug developers are becoming increasingly concentrated. Amidst the scaled expansion of other innovative pharmaceutical companies, orphan drug firms are continuously streamlining their pipeline portfolios to concentrate financial resources on the clinical advancement of a few lead candidates. Shire, the rare disease giant acquired by Takeda in 2018, has more than 50% of its clinical trials in Phase III/IV, a figure that stands at only around 30% for most pharmaceutical companies.
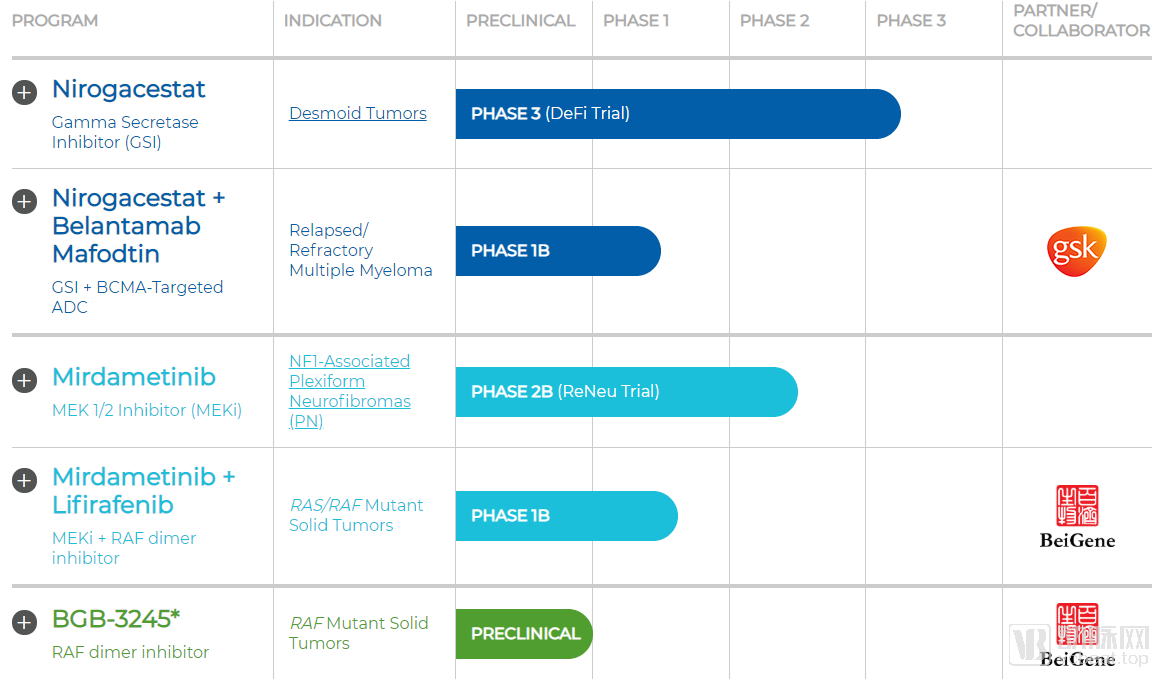
SpringWorks’ Pipeline in Development
SpringWorks Therapeutics has an extremely streamlined drug pipeline, with only two proprietary assets: nirogacestat and mirdametinib, both originally from Pfizer. Additionally, the company is collaborating with BeiGene on the development of BGB-3245, which was originally developed by BeiGene.
1.Nirogacestat:Desmoid Tumor Leader
Nirogacestat is unequivocally SpringWorks’ flagship drug. It has currently entered Phase III clinical trials for the treatment of patients with desmoid tumors. Nirogacestat has demonstrated favorable tolerability in previous clinical trials, and clinical efficacy has been observed in patients with desmoid tumors. In June 2018, the FDA granted Orphan Drug Designation to nirogacestat; in November, it approved the drug for Fast Track designation; and in August 2019, it further granted Breakthrough Therapy Designation.
The Phase III clinical trial of nirogacestat was initiated in April 2019 and is currently in the patient recruitment phase. According to information disclosed by SpringWorks Therapeutics in its prospectus, as long as nirogacestat demonstrates efficacy comparable to that observed in previous clinical trials during the Phase III study, the primary endpoint of the trial will be met. The trial is scheduled to last four years and is expected to conclude in April 2023.
Currently, there are no specific drugs available on the market for desmoid tumors. Nirogacestat is the drug with the most advanced clinical progress in the global desmoid tumor field, while the other two drugs targeting desmoid tumors are still in Phase I clinical trials. Patients with desmoid tumors account for only about 0.03% of all cancer patients, corresponding to approximately 5,000 new cases globally each year. Based on a treatment cost of $200,000 per patient and a conservative market penetration rate of 20%, Nirogacestat’s annual sales could reach $200 million.
2.Mirdametinib:Key Contenders Vying for the Neurofibroma Market
Mirdametinib is another orphan drug that SpringWorks inherited from Pfizer, originally coded as PD-0325901, for the treatment of neurofibromatosis type 1. Mirdametinib received FDA orphan drug designation and entered the Fast Track program in November 2018, and subsequently obtained orphan drug status from the European Commission in July 2019.
Mirdametinib just initiated a three-year Phase II clinical trial in July 2019. According to disclosures in SpringWorks’ prospectus, if the trial results are favorable, the company plans to directly apply for marketing approval in the United States based on the Phase II clinical trial data. Therefore, Mirdametinib may reach the market before Nirogacestat.
However, the market for neurofibromatosis is not as straightforward as that for glioblastoma. Multiple pharmaceutical companies have targeted the neurofibromatosis market, with Merck’s pembrolizumab, Roche’s bevacizumab, Novartis’s trametinib, and AstraZeneca’s selumetinib all undergoing research for neurofibromatosis and currently in Phase II clinical trials. Among these competitors, a significant number have obtained orphan drug designation for neurofibromatosis. Therefore, based on the current landscape, Mirdametinib does not hold a competitive advantage in the neurofibromatosis market. This may also be the key reason why SpringWorks Therapeutics aims to skip Phase III clinical trials and file for marketing approval early.
Although SpringWorks Therapeutics has not engaged in further product discussions with its former parent company, Pfizer, it has entered into a clinical research collaboration with another international pharmaceutical giant, GlaxoSmithKline (GSK). The partnership aims to evaluate the efficacy of combining Nirogacestat with GSK’s Belantamab in patients with relapsed/refractory multiple myeloma. Belantamab has already demonstrated therapeutic benefits in patients with multiple myeloma in Phase I/II clinical trials. This combination study will play a pivotal role in expanding the indications for Nirogacestat.
SpringWorks Therapeutics also maintains a close collaborative relationship with BeiGene. In June 2019, the two companies first announced the joint establishment of MapKure to develop BeiGene’s novel oncology drug, BGB3245. Subsequently, on September 6, they announced further collaboration to jointly initiate clinical trials evaluating the safety, tolerability, and preliminary efficacy of a combination therapy comprising BeiGene’s BGB-283 and SpringWorks’ PD-0325901.
Rare disease drug development has never been a market that attracts researchers in droves. The challenges of developing orphan drugs are no less than those for other pharmaceuticals, as they must undergo the entire drug development process—from molecular design and animal studies to clinical trials and post-marketing research. Failure at any stage can directly lead to the termination of a project. Even after overcoming numerous obstacles to achieve market approval, rare disease drugs face an extremely small patient population. This naturally lowers the “ceiling” for rare disease drugs compared to other therapeutics.
Even so, orphan drug R&D has still become one of the hottest niches in the pharmaceutical development sector. The reason lies in the powerful driving force of policies, where substantial policy incentives have expanded the profit margins within this small market.
1. Rapid market launch with low clinical trial costs
Regulatory agencies in various countries have established corresponding regulations to promote the research and development of orphan drugs. As early as the 1980s, the U.S. Food and Drug Administration (FDA) enacted the Orphan Drug Act. China’s latest revision of the Drug Administration Law explicitly incorporates support for the innovation of new drugs for rare diseases into its provisions and mandates priority review and approval for such drugs. Priority review reflects the high level of attention regulatory authorities pay to orphan drugs. The shortened approval timeline enables companies developing rare disease treatments to achieve profitability sooner, representing a key policy advantage for the orphan drug market.
Low clinical trial costs are another key factor making the rare disease drug development industry attractive. Phase III clinical trials for common diseases typically enroll 300–500 patients, whereas those for rare diseases require only around 100 participants. Although recruiting patients remains challenging despite their high proportion within the total patient population, the smaller enrollment size translates into comprehensive cost reductions for pharmaceutical companies—from drug supply expenses to trial management overhead—thereby helping them save significantly on R&D costs.
2. Market Exclusivity Period with Strong Policy Protection
In different countries and regions, regulatory authorities have granted orphan drugs extended periods of market exclusivity. During the market exclusivity period, generic versions of the protected drug will not be approved for marketing, and the launch of other drugs with the same indications will also be hindered, unless permission is obtained from the originator drug’s sponsor or the newly submitted drug is proven to demonstrate superior clinical efficacy. This period is 7 years in the United States, and 10 years in the European Union and Japan. China currently provides a 10-year data protection period for drugs used to treat rare diseases.
The establishment of an exclusivity period means that orphan drug developers gain a degree of market monopoly post-launch. This policy effectively compensates for the low return on investment resulting from the small market size of orphan drugs. Meanwhile, this policy has also created a “bottleneck” scenario for innovative drug development targeting the same indication. The first product to secure approval will directly attain a dominant position in the corresponding therapeutic area, significantly impacting the regulatory submissions and prospects of subsequent competing products.
3. Autonomous Pricing Driven by Patients' Essential Needs
Although the patient population for rare diseases is small, most rare diseases still lack established treatment options. This market gap means that orphan drugs approved first will become essential treatments for patients. Meanwhile, the FDA’s Orphan Drug Act grants pharmaceutical companies significant pricing autonomy. As a result, most orphan drugs currently on the market are priced extremely high. In May of this year, Novartis’ Zolgensma, a therapy for spinal muscular atrophy, just set a new record as the world’s most expensive drug at $2.125 million.
In reality, however, high prices may not lead to higher sales volumes. Drug pricing should be negatively correlated with the number of patients purchasing the medication. The core issue for rare-disease drug developers in setting prices is to determine an appropriate price point that maximizes their revenue.
UniQure’s Glybera serves as a cautionary tale in drug pricing. Approved by the European Union in 2012 and officially launched in 2014, Glybera was indicated for the treatment of lipoprotein lipase deficiency. As the first approved gene therapy, it was also the most expensive drug at the time, with a treatment price of €1.1 million that far surpassed all other medications. However, patients appeared reluctant to embrace it. During its four years on the market, only one patient received Glybera treatment, and this was made possible only through substantial reimbursement from an insurance company. In April 2017, as its marketing authorization was nearing expiration, UniQure announced that it would not seek renewal, marking the quiet exit of the first-ever gene therapy from the stage.
Real-world studies, which have recently gained significant traction, have established clear applications in the field of rare diseases. Relevant policies issued in China have explicitly incorporated the use of real-world evidence to support the research and development of therapeutic drugs for rare diseases as well as regulatory decision-making. Due to the challenges in patient recruitment for clinical trials of rare diseases, real-world data derived from natural disease cohorts can serve as external controls for non-randomized, single-arm trials. Furthermore, real-world studies can accelerate patient recruitment for clinical trials by analyzing patient distribution patterns.
Molecular Subtyping of Diseases Is Creating New Opportunities for Indication Expansion in Rare Disease Drugs. Supported by scientific research and molecular diagnostics, our understanding of disease etiology is increasingly deepening at the molecular level. Identical molecular alterations may lead to different diseases under varying internal environments. Conditions sharing consistent molecular subtypes can be treated with drugs targeting the same therapeutic target.
Orphan drugs are, in essence, still targeted therapies. They have the opportunity to identify other indications driven by the same target within new molecular subtypes of diseases. Leveraging the aforementioned advantages of orphan drugs, both the market approval process and clinical trial timelines are significantly shortened. Therefore, after rapid market entry, these drugs can undergo post-marketing studies as approved products to explore potential additional indications. Obtaining approval for new indications of an already marketed drug is considerably easier than approving a new drug. Consequently, for target-related drugs that impact multiple disease areas, securing initial approval as an orphan drug and subsequently researching other indications represents a viable and strategic option.