NMPA Releases Technical Guideline on Clinical Trial Endpoints for Advanced Non-Small Cell Lung Cancer, Setting New Standards for Drug Development
Yesterday, the National Medical Products Administration (NMPA) officially released the “Technical Guidance on Clinical Trial Endpoints for Advanced Non-Small Cell Lung Cancer,” marking the end of the wait since theOnly three months have passed since the release of the “Technical Guidelines for Clinical Trial Endpoints in Advanced Non-Small Cell Lung Cancer (Draft for Comment).”The officially released version is largely consistent with the previous draft for comments in terms of thematic content, with modifications primarily made to certain details.
Technical Guidelines for Clinical Trial Endpoints in Advanced Non-Small Cell Lung Cancer
I. Preface
Lung cancer ranks first globally and in China in terms of both incidence and mortality among malignant tumors [1]. In 2015, there were 3.929 million new cancer cases and 2.338 million cancer-related deaths in China, including 787,000 new lung cancer cases and 631,000 lung cancer deaths [2]. Non-small cell lung cancer (NSCLC) accounts for 85% of all lung cancers, with the majority of patients diagnosed at an advanced stage. In recent years, the application of small-molecule tyrosine kinase inhibitors (TKIs), anti-angiogenic agents, and immune checkpoint inhibitors has significantly improved patient survival.
Advanced non-small cell lung cancer (NSCLC) has become a focal area for the development of novel antineoplastic agents. With a plethora of innovative drugs emerging, the chain of clinical evidence is becoming increasingly complex, giving rise to intricate endpoints and study designs—including surrogate endpoints, intermediate clinical endpoints, and other innovative endpoints. Complex designs such as parallel testing of co-primary endpoints and sequential testing of composite endpoints have also emerged. Existing guidelines are not yet comprehensive enough to cover these developments. Therefore, this article aims to elucidate general considerations for the design and regulatory review of endpoints in clinical trials for advanced NSCLC. It is intended to provide reference for antineoplastic drug developers in designing clinical trials and selecting endpoints for advanced lung cancer, thereby facilitating the scientific and efficient determination of drug efficacy, enhancing clinical development efficiency, and enabling patients to benefit earlier.
This guideline applies to the design of clinical trials and selection of endpoints for registration purposes in support of indications for advanced non-small cell lung cancer (NSCLC). The design of clinical trials for antineoplastic agents covered herein shall also comply with general principles of clinical trial design, including but not limited to the International Council for Harmonisation of Technical Requirements for Pharmaceuticals for Human Use (ICH) guidelines E8, E9, E10, and E17, as well as relevant provisions issued by the National Medical Products Administration (NMPA), such as the “Technical Guideline on Clinical Trial Endpoints for Antineoplastic Agents” and the “Technical Guideline on Clinical Trials for Antineoplastic Agents.”
The views presented in this guideline reflect the current understanding of the National Medical Products Administration (NMPA) regarding clinical trial design and endpoint selection for advanced non-small cell lung cancer (NSCLC), and do not cover all scenarios encountered in the development of novel anti-tumor drugs. R&D professionals are encouraged to explore scientifically innovative endpoints and trial designs, and to engage in timely communication and exchange with the NMPA’s review divisions.
II. Background
The current treatment goals for advanced non-small cell lung cancer (NSCLC) are to prolong survival and improve quality of life. The endpoints in pivotal registration trials should effectively reflect metrics or events indicative of clinical benefit. Prior to the 1990s, most approved anticancer drugs used overall survival (OS) as the primary endpoint. OS is clearly defined, objective, and robust, serving as the gold standard for reflecting patient survival benefits; however, trials based on OS are time-consuming and require large sample sizes. To accelerate drug approval and improve treatment accessibility, regulatory authorities have implemented accelerated approval pathways for drugs demonstrating substantial clinical benefit in the context of refractory diseases. This approach allows the use of surrogate endpoints that can reasonably predict clinical benefit, such as objective response rate (ORR) and progression-free survival (PFS), as primary endpoints to support the approval of new drugs [3-5].
In recent years, the treatment paradigm for advanced non-small cell lung cancer (NSCLC) has shifted from cytotoxic chemotherapy based on histopathology to molecular targeted therapy guided by predictive biomarkers of efficacy, such as driver gene mutations, as well as combination therapies involving immunotherapy, cytotoxic agents, and anti-angiogenic drugs. Regulatory authorities typically approve new drugs based on direct clinical benefits, such as prolonged overall survival (OS) and improvement in tumor-related symptoms. Alternatively, surrogate endpoints that predict clinical benefit, such as higher objective response rates (ORR) and sufficient duration of response (DOR), may also be accepted for drug approval. Regulatory agencies need to balance the impact of surrogate endpoints and innovative study designs on the assessment of clinical benefit. This guideline will discuss the general regulatory requirements for efficacy evaluation and the selection of endpoints to support approval.
III. Common Endpoints in Clinical Trials of Antineoplastic Drugs
Commonly used endpoints in clinical trials of antitumor drugs can be categorized into three types based on their sources: endpoints based on death events, such as overall survival (OS) and OS rate; endpoints based on tumor measurements, such as objective response rate (ORR) assessed by the Response Evaluation Criteria in Solid Tumors (RECIST), or time to progression (TTP), progression-free survival (PFS), and time to treatment failure (TTF) based on RECIST and visit schedules; and endpoints based on symptom assessment, such as patient-reported outcomes (PROs) including pain relief and quality of life (QoL).
The selection of endpoints should be comprehensively considered in conjunction with factors such as tumor staging, prior treatments, and characteristics of therapeutic onset.
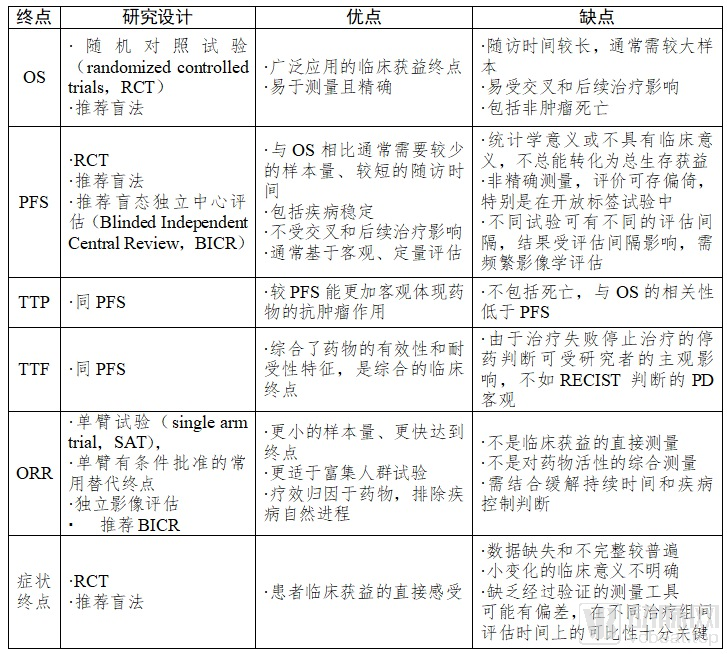
Table 1. Comparison of Common Clinical Trial Endpoints for Drugs in Advanced NSCLC
(I) Endpoints Based on Mortality Events
OS is defined as the time from randomization to death from any cause. The determination of OS is precise and reliable, less prone to bias, and it is often used as the primary endpoint. Clinical trials with OS as the primary endpoint require a randomized controlled design, often necessitating larger sample sizes and longer follow-up periods, and are susceptible to the effects of crossover and subsequent treatments.
Overall Survival (OS) rate is defined as the proportion of subjects surviving within the same trial arm from randomization to a specified time point. As an intermediate clinical endpoint for OS, it has served as a secondary endpoint in previous studies. In randomized clinical trials, treatment benefits can be observed by comparing OS rates, such as the 1-year OS rate. Although the calculation of OS rate is precise and reliable and can be achieved earlier, it is typically used as a descriptive endpoint. The assessment of OS rate is heavily influenced by the choice of time point, and its clinical and statistical significance remains unclear. Premature analysis of OS rate may lead to unblinding, which is detrimental to the evaluation of survival benefits. Currently, OS rate is primarily utilized as a secondary endpoint and supportive evidence.
(II) Endpoints Based on Tumor Measurements
Clinical decision-making in oncology is often based on imaging assessments of lesions, with tumor measurement endpoints considered indicative of clinical benefit. The Response Evaluation Criteria in Solid Tumors (RECIST), currently version 1.1, is widely used to evaluate treatment efficacy in non-small cell lung cancer (NSCLC). With the adoption of immune checkpoint inhibitors, the immune response evaluation criteria in solid tumors (iRECIST) have begun to be applied in clinical trials; however, iRECIST is not suitable for assessing progression-free survival (PFS) when the control arm involves standard chemotherapy. In clinical trials of cancer immunotherapy aimed at new drug registration, RECIST 1.1 remains the most commonly used criterion for determining tumor response. It is recommended to incorporate iRECIST alongside traditional RECIST assessments and compare their results in clinical trials involving immunotherapy alone or using a “head-to-head” design.
ORR is defined as the proportion of patients whose tumor volume shrinks to a pre-specified value and remains so for a minimum required duration. ORR is the sum of the proportions of complete response (CR) and partial response (PR). ORR excludes stable disease (SD), thereby eliminating the influence of the natural course of the disease. Compared with the disease control rate (DCR), ORR provides a more reliable reflection of a drug’s antitumor activity and is a commonly used surrogate endpoint in single-arm clinical trials.
DOR is defined as the time from the first assessment of objective response to the first assessment of progressive disease (PD) or death from any cause prior to PD, reflecting the duration of the overall response rate (ORR).
PFS is defined as the time from randomization to objective tumor progression or death from any cause, serving as a surrogate endpoint for OS. Compared with TTP, PFS includes deaths from any cause, demonstrates a higher correlation with OS, and is not influenced by subsequent treatments, making it the most commonly used surrogate endpoint in randomized controlled clinical trials.
TTP is defined as the time from randomization to the occurrence of objective tumor progression, excluding death. While TTP can accurately reflect short-term survival benefits derived from treatment, its correlation with clinical benefit is inferior to that of PFS and TTF due to the exclusion of death.
Time to Treatment Failure (TTF) is defined as the time from randomization to treatment failure or withdrawal from the trial. Reasons for withdrawal may include patient request, disease progression, death, or adverse events. Compared with Progression-Free Survival (PFS), TTF captures withdrawals not caused by disease progression and may include continued treatment after disease progression, making it a comprehensive clinical endpoint. However, because TTF does not adequately distinguish between factors such as drug efficacy and tolerability, it is currently not commonly used as the primary endpoint in confirmatory studies of anticancer drugs.
The determination of TTP, PFS, and TTF outcomes is influenced by visit interval design and trial quality; excessive censoring and truncation arising from loss to follow-up or the absence of observed endpoint events during the study period will compromise the analysis of these endpoints.
Considerations for Patient Inclusion and Endpoints in Lung Cancer with Brain Metastases
Brain metastases are a significant cause of disease progression and treatment failure in advanced non-small cell lung cancer (NSCLC), necessitating clinical intervention for symptomatic patients. Previous clinical trials in advanced NSCLC have largely excluded patients with brain metastases or enrolled only those with asymptomatic, stable brain metastases following local and systemic therapies. Consequently, the trial results fail to reflect the efficacy of drugs in patients with brain metastases, leading to a lack of critical effectiveness data. Given the high incidence of brain metastases in lung cancer and the importance of controlling them as a key therapeutic goal in metastatic NSCLC, it is encouraged to include patients with brain metastases in clinical trials based on preliminary clinical study results of the drug. Attention should be paid to collecting complete and accurate baseline information on brain metastases, and incorporating assessments of local intracranial benefit into efficacy evaluations, such as intracranial objective response rate, duration of intracranial response, and time from the appearance of new brain metastases to intracranial disease progression.
(3) Endpoints Based on Symptom Assessment
Improvement in symptoms and signs among cancer patients is considered a direct clinical benefit rather than a surrogate endpoint. Regulatory authorities may approve new drugs based on significant symptom improvement, such as control of malignant effusion, alleviation of cancer-related fatigue, and reduction in skeletal-related events. Different symptom indicators within a composite symptom endpoint should have similar clinical importance. The clinical benefit should not be attributed solely to the improvement of individual indicators; therefore, specific individual indicators should be analyzed when evaluating the composite endpoint.
When improvement in symptoms and signs is used as the primary endpoint to support the approval of antineoplastic drugs, it should be possible to distinguish whether the improvement is due to alleviation of tumor-related symptoms or to reduced or absent toxicity of the investigational drug. If relief of a specific symptom is selected as the endpoint, participants must have presented with that symptom at baseline, and the symptom must be attributable to the disease.
Missing data and inadequate assessment will increase the complexity of evaluating symptom endpoints. The visit schedule should be strictly implemented to balance and maximize the completion rate of visits. The statistical analysis plan (SAP) should specify how missing data will be handled, and information suitable for analysis should continue to be collected when patients discontinue treatment. Prospective data collection on multiple symptoms should be conducted. In cases involving multiple testing, the SAP must describe the necessary methods for controlling Type I error.
IV. Design and Endpoint Considerations for Registration Clinical Trials of New Drugs in Advanced Non-Small Cell Lung Cancer
In China, advanced non-small cell lung cancer (NSCLC) exhibits a high prevalence of driver gene mutations. Clinical trial designs for this condition can be categorized into enriched population trials and non-enriched population trials, based on the presence or absence of predictive biomarkers of therapeutic efficacy.
(I) Clinical Trials Enriching Populations with Tumor Biomarkers
Trial designs for populations enriched with tumor biomarkers should consider factors such as biomarkers and lines of therapy:
1. Biomarkers
Biomarker Selection: Specific biomarker nomenclature must be clearly defined; for example, it should be specified that a given epidermal growth factor receptor-tyrosine kinase inhibitor (EGFR-TKI) is indicated for EGFR exon 19 deletion mutations and exon 21 L858R mutations. The nomenclature of biomarkers associated with advanced lung cancer shall comply with ICH E15.
Requirements for Biological Specimens: Factors such as specimen collection, fixation, and storage and transport conditions can significantly affect the interpretation of results. It is necessary to clearly define the acceptable sources of biomarkers for the study, such as primary tumors, metastatic lesions, or peripheral blood (e.g., ctDNA); specify the fixation requirements, such as formalin-fixed paraffin-embedded (FFPE) or fresh-frozen tissues; and clarify the storage and transport conditions. These details should be described in the study protocol or further elaborated into corresponding standard operating procedures (SOPs). The general principles for the collection, processing, transportation, storage, and disposal of biological specimens shall comply with the relevant requirements of ICH E18.
Companion Diagnostic Method: After determining the biomarker and specimen requirements, the companion diagnostic method must be clearly defined.
Some widely used companion diagnostics with proven efficacy for biomarkers, such as sequencing or the amplification refractory mutation system (ARMS) assay for detecting EGFR sensitizing mutations, can be performed in local laboratories without requiring specific companion diagnostic/diagnostic kits.
Companion diagnostics for biomarkers that are not yet widely used and are still in clinical development, such as PD-L1 expression, c-MET amplification, and high microsatellite instability/DNA mismatch repair deficiency (MSI-H/dMMR), require clearly defined companion diagnostic methods or diagnostic kits, with the use of central laboratories recommended (local central laboratories are acceptable).
Encourage the concurrent development of new drugs and in vitro companion diagnostic reagents. If a novel biomarker or assay method is developed, sufficient validation data for the selected biomarker should be submitted with the marketing authorization application. It is recommended to engage in regulatory discussions regarding submission requirements before initiating pivotal registration clinical trials. Submission materials should be prepared in accordance with the background, structure, and format requirements outlined in ICH E16, with progress made in a stepwise manner.
2. Considerations for Line of Therapy: Typically, treatment-naïve patients have a better performance status and superior tolerability compared to those who have undergone multiple prior lines of therapy. Therefore, stratification by the number of prior lines of therapy should be considered in the trial design.
The decision to support new drug registration with either randomized controlled trials (RCTs) or single-arm trials (SATs) should be made by comprehensively considering factors such as disease background, drug accessibility, clinical needs, and prior clinical trial data.
3. Design and Endpoint Considerations for RCTs: Randomized controlled trials (RCTs) are the most reliable method for confirming drug efficacy. Overall survival (OS) is the gold standard for reflecting the clinical benefit of antitumor drugs and is typically used as the primary endpoint in RCTs. With the diversification of treatment options, the continuous prolongation of OS has increased the difficulty of evaluation; therefore, progression-free survival (PFS) alone or co-primary endpoints of PFS and OS are acceptable as primary endpoints in registration studies for previously untreated advanced non-small cell lung cancer (NSCLC). For RCTs enrolling patients with relapsed/refractory disease or those lacking standard treatment options, OS remains the recommended primary endpoint.
While selecting the primary endpoint, the trial design—superiority, non-inferiority, or equivalence—should also be determined. When the control group receives standard therapy, superiority, non-inferiority, or equivalence designs are acceptable; if an equivalence or non-inferiority hypothesis is chosen, the applicant must communicate with regulatory authorities regarding the equivalence or non-inferiority margins. In the case of placebo-controlled trials or add-on designs, only superiority designs are accepted. For placebo-controlled clinical trials, it is recommended to combine with best supportive care (BSC) to enhance subject protection.
In randomized controlled trials (RCTs), regulatory agencies encourage the use of reasonable surrogate endpoints and prespecified interim analyses to support approval. In cases involving multi-arm RCTs, prespecified interim analyses, co-primary endpoints, or multiple testing of primary endpoints, it is essential to maintain the overall family-wise Type I error rate for the primary endpoint(s) under complex designs at a two-sided level of less than 0.05.
4. Design and Endpoint Considerations for SATs: When a new drug demonstrates breakthrough efficacy in the context of refractory diseases, applicants may communicate with regulatory authorities to consider using a Single-Arm Trial (SAT) with surrogate endpoints as the pivotal registration study supporting conditional approval, with full approval to be obtained through subsequent confirmatory randomized controlled trials.
The greatest limitation of single-arm trials (SATs) is the bias arising from the lack of a control group, which leads to uncertainty in the evaluation of drug efficacy. Therefore, if an SAT is considered as a pivotal study to support the accelerated approval of a new drug, it should meet the following conditions:
Sufficient Historical Data: Historical control data for SAT should be derived from high-level evidence-based medicine sources, characterized by similar time periods, identical disease backgrounds, and adequate sample sizes. Such data may originate from individual randomized controlled trials (RCTs), real-world evidence, systematic reviews, and meta-analyses, with an assessment of the reliability of these data in the current context.
Background of Refractory Diseases: SAT should be conducted in recurrent and refractory diseases with urgent unmet medical needs. Subjects enrolled in SAT should be patients who have failed current standard treatments or for whom no standard treatment is available.
Primary Endpoints Based on Response Rate/Objective Metrics: Due to the lack of a control group and the uncertainty associated with historical controls, single-arm trials (SATs) must carefully consider the primary study endpoint. The objective response rate (ORR) is precisely defined, excludes the natural history of the tumor, and directly reflects the antitumor efficacy of the drug. Therefore, ORR is commonly adopted as the primary endpoint in SATs for advanced non-small cell lung cancer (NSCLC).
Preset Target Value for Response Rate: A target value for the Objective Response Rate (ORR) should be preset. The lower bound of the 95% confidence interval for the target ORR should be determined through communication with regulatory authorities, based on sufficient historical data combined with the product’s own early clinical data. It is generally expected that the target ORR will show a substantial improvement compared with historical controls; specifically, the lower bound of the 95% confidence interval for the target ORR must exceed the upper limit of the historical data for standard therapy [10].
Duration of Response: On the basis of confirming that the objective response rate (ORR) has met the predefined criteria, it is crucial to evaluate the duration of response. A sustained duration of response enhances the robustness of therapeutic benefit, while a longer treatment exposure also reflects information on the drug’s safety and tolerability.
Independent Review Committee (IRC): The SAT must utilize an independent imaging review committee to mitigate investigator bias, and employ the objective response rate (ORR) assessed by investigators for sensitivity analysis.
Requirements and Models for Post-Marketing Approval of SATs: As SATs are granted conditional accelerated approval, applicants must communicate with regulatory authorities regarding the conditions for full approval before initiating SAT studies. Typically, randomized controlled trials (RCTs) conducted in patients within the same line of therapy are regarded as rigorous confirmatory clinical trials [11]. Since molecular targeted therapies do not strictly emphasize chemotherapy line distinctions, confirmatory clinical trials with robust statistical hypotheses conducted in prior-line patient populations may also be accepted. Regulatory authorities will comprehensively consider specific requirements for confirmatory studies, taking into account factors such as the incidence rate of the target disease population, the strength of efficacy data, and ethical considerations. In rare cases where RCTs cannot be completed, real-world data may be used to support full approval.
(II) Clinical Trials Not Enriching the Population Based on Tumor Biomarkers
In advanced NSCLC, due to the lack of established predictive biomarkers for efficacy, clinical trials of cytotoxic and anti-angiogenic agents are often conducted in unselected populations (with consideration of histological stratification), and some immune checkpoint inhibitors have also been studied in unselected populations.
Non-enrichment trials should focus on prognostic factors (such as histopathological type, ECOG performance status score, prior treatments, and baseline brain metastases) and stratify important prognostic factors. In non-enrichment trials, the line of therapy is a key basis for selecting the primary endpoint.
The current first-line standard of care for advanced non-small cell lung cancer (NSCLC) without driver gene mutations is platinum-based doublet chemotherapy, which may be combined with anti-angiogenic therapy or immunotherapy. Similar to randomized controlled trial (RCT) designs in enriched populations, RCTs for first- or second-line treatment in unselected populations may select progression-free survival (PFS) as the primary endpoint. For studies beyond the second line, overall survival (OS) is recommended as the primary endpoint; however, the potential impact of subsequent therapies and crossover must be considered. The efficacy of immune checkpoint inhibitors is primarily reflected in long-term survival benefits for a subset of patients. Therefore, evaluation of OS benefit should be emphasized in first-line treatment, and it is recommended to designate both OS and PFS as co-primary endpoints, or OS as the sole primary endpoint. Given that the efficacy of immune checkpoint inhibitors generally exhibits a delayed separation and tailing effect in survival curves, interim analyses are not recommended when the number of events is insufficient (e.g., less than 50% of the planned number of OS events).
The purpose of maintenance therapy is to consolidate the benefits of first-line treatment and prolong the time to disease progression. Therefore, the primary endpoints of clinical trials for maintenance therapy should align with the treatment objectives; PFS or TTF may be selected as the primary endpoint. When the efficacy of maintenance therapy translates into long-term survival benefits, OS should be chosen as the primary endpoint.
Apart from considerations of biomarkers, non-enriched and enriched populations follow the same trial design and endpoint selection.
V. Considerations for Primary Endpoint Benefits in Registration Clinical Trials of New Drugs for Advanced Non-Small Cell Lung Cancer
In current drug development for advanced non-small cell lung cancer (NSCLC), randomized controlled trial (RCT) designs with different study endpoints evaluate clinical value after achieving statistical significance in the primary endpoint.
(I) Overall Survival
At the current stage, the treatment goals for advanced non-small cell lung cancer (NSCLC) are to prolong patient survival and improve quality of life. Overall survival (OS) is a direct endpoint reflecting survival benefit, and its clinical significance outweighs that of surrogate endpoints such as progression-free survival (PFS). Subsequent therapy is the most significant factor influencing OS; therefore, assuming balanced subsequent treatments, experts both domestically and internationally currently consider that an OS benefit of more than 2.5 months in metastatic squamous cell lung cancer, and more than 3.25 months in non-squamous NSCLC, holds significant clinical value, with the point estimate of the target hazard ratio (HR) typically not exceeding 0.80 [12]. The hazard ratio and median benefit are usually evaluated in combination, with both metrics meeting these thresholds considered indicative of a more robust OS benefit.
(II) Progression-Free Survival
PFS serves as a surrogate endpoint for OS, and it remains uncertain whether PFS benefits can translate into OS benefits. Various non-treatment factors can lead to statistically significant differences in PFS, such as imbalanced prognostic factor stratification, selection of weaker control treatments, and even the design of efficacy assessment time points. At present, in first-line treatment for advanced NSCLC, a PFS benefit of more than 3 months in lung squamous cell carcinoma and more than 4 months in non-squamous cell carcinoma holds significant clinical value, with the point estimate of the target hazard ratio (HR) typically no higher than 0.70 [12].
(3) Objective Response Rate
When ORR is the primary endpoint, the core consideration is whether the ORR assessed by IRC in the full analysis set has reached the pre-specified target value, while also ensuring sufficient duration of response. In the current context of advanced NSCLC, the lower bound of the 95% confidence interval for ORR must meet the pre-specified target value.
VI. Conclusion
Advanced non-small cell lung cancer (NSCLC) is a focal point for the development of antineoplastic agents. With the advent of new drugs, the body of evidence supporting the treatment of advanced NSCLC is increasingly robust, and the design of clinical trials and selection of endpoints have become more complex. At present, prolonging survival time and improving quality of life remain the core objectives in the treatment of advanced lung cancer. The selection of clinical trial endpoints is guided by the principle of objectively and efficiently reflecting the clinical benefits of tumor therapy. Scientific advancements will drive the development of antineoplastic products, encouraging sponsors to communicate with regulatory authorities and explore innovative trial designs and study endpoints. This guidance will be updated in due course based on progress in clinical trials for advanced NSCLC.
References
[1] Siegel RL,Miller KD,Jemal A. Cancer statistics,2016. CA Cancer J Clin. 2016. 66(1): 7-30.
[2] Zheng R, Sun K, Zhang S, et al. Analysis of the epidemiology of malignant tumors in China in 2015. Chinese Journal of Oncology, 2019, 41(1): 19-28.
[3] Downing NS,Aminawung JA,Shah ND,Braunstein JB,Krumholz HM,Ross JS. Regulatory review of novel therapeutics--comparison of three regulatory agencies. N Engl J Med. 2012. 366(24): 2284-93.
[4] Carpenter D,Zucker EJ,Avorn J. Drug-review deadlines and safety problems. N Engl J Med. 2008. 358(13): 1354-61.
[5] Greene JA,Podolsky SH. Reform,regulation,and pharmaceuticals--the Kefauver-Harris Amendments at 50. N Engl J Med. 2012. 367(16): 1481-3.
[6] Nishino M,Giobbie-Hurder A,Gargano M,Suda M,Ramaiya NH,Hodi FS. Developing a common language for tumor response to immunotherapy: immune-related response criteria using unidimensional measurements. Clin Cancer Res. 2013. 19(14): 3936-43.
[7] Seymour L,Bogaerts J,Perrone A,et al. iRECIST: guidelines for response criteria for use in trials testing immunotherapeutics. Lancet Oncol. 2017. 18(3): e143-e152.
[8] Lin NU,Lee EQ,Aoyama H,et al. Response assessment criteria for brain metastases: proposal from the RANO group. Lancet Oncol. 2015. 16(6): e270-8.
[9] Reck M,Rodríguez-Abreu D,Robinson AG,et al. Pembrolizumab versus Chemotherapy for PD-L1-Positive Non-Small-Cell Lung Cancer. N Engl J Med. 2016. 375(19): 1823-1833.
[10] Khozin S,Weinstock C,Blumenthal GM,et al.Osimertinb for the treatment of metastatic EGFR T790M mutation-positive non-small cell lung cancer.Clin Cancer Res.2017,23(9):2131-35.
[11] Odogwu L,Mathieu L,Goldberg KB,et al.FDA benefit-risk assessment of osimertinib for the treatment of metastatic non-small cell lung cancer harboring epidermal growth factor receptor for T790M mutation.Oncologist.2018,23(3):353-59.
[12] Ellis LM,Bernstein DS,Voest EE,et al. American Society of Clinical Oncology perspective: Raising the bar for clinical trials by defining clinically meaningful outcomes. J Clin Oncol. 2014. 32(12): 1277-80.