JunLian Healthcare Global Drug Pipeline Update - Issue 23
Sep 23, 2019 21:53
CST Updated
21:53

Legend Capital
Early-stage venture capital and growth-stage private equity investment institutions

❖New Drug Data for This Week: A Total of10 in total, including 6 oncology drugs, and one each for metabolic, immune, allergic, and CNS diseases
❖The FDA has officially approved Novo Nordisk’s New Drug Application for Rybelsus (oral semaglutide, once daily), based on results from ten PIONEER clinical trials. In head-to-head studies, Rybelsus outperformed sitagliptin, empagliflozin, and liraglutide, cementing its dominant market position—a feat reminiscent of Lipitor’s historic industry dominance. In the current landscape of diabetes mellitus (DM) management, a product with glucose-lowering effects alone is no longer competitive; the focus has shifted to additional benefits such as weight reduction and cardiovascular outcomes. With a powerhouse like Rybelsus in the market, it has become exceedingly difficult for biotech companies to enter this therapeutic area.
❖The 2019 CSCO Annual Meeting was held in Xiamen, Fujian Province, from September 18 to 22. Overall, as much of the data had already been previewed or leaked, there were no particularly stunning or groundbreaking revelations. The main highlights of the conference focused on immunotherapy (PD-1 inhibitors) and precision therapy for lung cancer. Notable presentations included: pembrolizumab monotherapy improving overall survival (OS) in Chinese patients with PD-L1-positive non-small cell lung cancer (NSCLC); camrelizumab combined with chemotherapy demonstrating significant benefits as first-line treatment for non-squamous NSCLC; Pfizer’s second-generation EGFR TKI dacomitinib attempting to challenge osimertinib by leveraging its OS advantage; and Betta Pharmaceuticals’ ensartinib challenging Pfizer’s second-generation ALK inhibitor crizotinib, based on its median progression-free survival (PFS) and central nervous system (CNS) efficacy benefits.
US FDA Approves Novo Nordisk’s Oral Semaglutide for Marketing to Improve Glycemic Control in Patients with Type 2 Diabetes
U.S. FDA Approves Novo Nordisk’s Oral Semaglutide for Marketing, to Improve Glycemic Control in Patients with Type 2 Diabetes When Combined with Diet and Exercise
Once-daily oral semaglutide tablets are the first FDA-approved oral glucagon-like peptide-1 receptor agonist (GLP-1 RA).
The application for marketing authorization and the approval of oral semaglutide were based on the results of 10 PIONEER clinical trials, involving a total of 9,543 patients with type 2 diabetes.
A series of “head-to-head” clinical trials in the PIONEER program have separately confirmed that oral semaglutide offers significant advantages over sitagliptin, empagliflozin, and liraglutide in reducing HbA1c levels and promoting weight loss. Moreover, oral semaglutide demonstrates a safety and tolerability profile consistent with other GLP-1 receptor agonists, with nausea being the most common adverse event.
Semaglutide'sPioneer Series Research
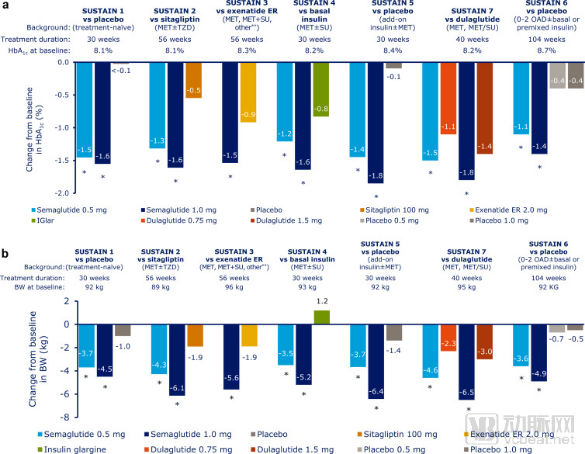
Data Source:Science direct
Novartis’s Fully Human Anti-IL-17A Monoclonal Antibody Meets Primary and All Secondary Endpoints in Phase 3 Clinical Trial for Non-Radiographic Axial Spondyloarthritis
Novartis’s fully human anti-IL-17A monoclonal antibody Cosentyx (secukinumab) met the primary endpoint and all secondary endpoints in the Phase 3 PREVENT trial for non-radiographic axial spondyloarthritis.
Cosentyx is the first fully human biologic agent that directly inhibits IL-17A.
555 adult patients with non-radiographic axial spondyloarthritis participated in this 2-year, randomized, double-blind, placebo-controlled Phase 3 clinical study.
Compared with placebo, patients treated with Cosentyx demonstrated a statistically significant and clinically meaningful reduction in disease activity. At 16 weeks, the proportion of patients achieving Assessment of SpondyloArthritis International Society 40 (ASAS40) response criteria was significantly higher than that in the control group, thereby meeting the primary endpoint of the trial.
U.S. FDA Approves Merck’s Keytruda in Combination with Eisai’s Lenvatinib for the Treatment of Patients with Specific Advanced Endometrial Cancer
The U.S. FDA Approves Merck’s Keytruda in Combination with Eisai’s Lenvatinib for the Treatment of Patients with Specific Advanced Endometrial Cancer Who Are Not Microsatellite Instability-High (MSI-H) or Mismatch Repair Deficient (dMMR)
Keytruda is a PD-1 inhibitor, and lenvatinib is a tyrosine kinase receptor inhibitor.
In a single-arm Phase II clinical trial, 108 patients with metastatic endometrial cancer were enrolled, of whom 87% (94 patients) had tumors that were not MSI-H or dMMR.
Among these 94 patients, the combination of Keytruda and Lenvima achieved an objective response rate of 38.3% (95% CI, 29%-49%), with a complete response rate of 10.6% and a partial response rate of 27.7%.
Janssen Announces U.S. FDA Approval of Erleada (apalutamide) for the Treatment of Metastatic Castration-Sensitive Prostate Cancer (mCSPC)
Janssen, a subsidiary of Johnson & Johnson, announced that the U.S. FDA has approved Erleada (apalutamide) for the treatment of metastatic castration-sensitive prostate cancer (mCSPC).
Apalutamide (阿帕他胺) is a second-generation nonsteroidal androgen receptor (AR) inhibitor.
In a phase 3, randomized, placebo-controlled, double-blind study, 1,052 patients from 23 countries were enrolled. The trial allowed patients to have received prior treatment for localized prostate lesions and docetaxel (without disease progression) before enrollment.
Compared with placebo + ADT, Erleada + ADT significantly prolonged OS, reducing the risk of death by 33% (HR = 0.67; 95% CI, 0.51–0.89; P = 0.0053).
FDA-Convened Advisory Committee on Allergenic Products Supports AR101, Developed by Aimmune Therapeutics, for the Treatment of Peanut Allergy in Children and Adolescents
Aimmune Therapeutics Announces FDA-Convened Allergic Products Advisory Committee (APAC) Supports AR101 (Proposed Trade Name Palforzia) for the Treatment of Peanut Allergy in Children and Adolescents
The mechanism of Palforzia involves administering small amounts of peanut protein to patients with peanut allergy, thereby gradually inducing desensitization of the patient’s immune system to peanut protein.
In the results of multiple Phase III clinical trials, more than 1,000 patients participated in these clinical trials.
Patients receiving Palforzia were able to tolerate significantly higher doses of peanut protein than those in the placebo group at the end of the clinical trials, with a 100-fold increase in the median tolerated protein dose.
The U.S. FDA Has Accepted the Biologics License Application for Seattle Genetics’ Antibody-Drug Conjugate for the Treatment of Urothelial Carcinoma
Seattle Genetics and Astellas Pharma jointly announced that the U.S. FDA has accepted their Biologics License Application (BLA) for enfortumab vedotin, an antibody-drug conjugate (ADC) co-developed by the two companies, for the treatment of patients with locally advanced or metastatic urothelial cancer.
Enfortumab vedotin is an antibody-drug conjugate.
In the pivotal Phase 2 clinical trial, a total of 128 patients who had previously received PD-1/L1 inhibitor therapy and platinum-based chemotherapy in the neoadjuvant/adjuvant setting were enrolled.
Enfortumab vedotin achieved an objective response rate (ORR) of 44% in these patients, with 12% achieving a complete response (CR).
Acadia Pharmaceuticals Announces Pimavanserin Met Primary Endpoint in Phase 3 Trial for the Treatment of Dementia-Related Psychosis
Acadia Pharmaceuticals Announces That Pimavanserin, for the Treatment of Dementia-Related Psychosis, Met the Primary Endpoint in the Pivotal Phase 3 HARMONY Trial
Pimavanserin is a selective serotonin inverse agonist (SSIA) that selectively targets the 5-HT2A receptor.
In the Phase 3 HARMONY trial, patients who met the prespecified remission criteria during the 12-week open-label stabilization period were randomized to either continue receiving pimavanserin or switch to placebo in the subsequent double-blind phase.
Compared with placebo, pimavanserin significantly prolonged the duration of remission before recurrence of dementia-related psychosis in patients, thereby meeting the primary endpoint of the trial.
Innovent Biologics’ IBI301 Meets Primary Endpoints in Two Phase 3 Trials
Innovent Biologics conducted a pharmacokinetic (PK) comparative study of IBI301 in patients with CD20-positive B-cell lymphoma who had achieved complete response after prior treatment, as well as a clinical efficacy comparative study in previously untreated patients with CD20-positive diffuse large B-cell lymphoma (DLBCL). The Phase 3 results demonstrated that the primary endpoint was met.
IBI301 is a rituximab biosimilar, an anti-CD20 monoclonal antibody.
A pharmacokinetic (PK) comparative study of IBI301 versus the originator rituximab injection, conducted in patients with CD20-positive B-cell lymphoma who achieved complete response (CR) after treatment, enrolled a total of 181 subjects, with 89 in the IBI301 group and 92 in the originator drug group.
IBI301 demonstrates equivalent clinical efficacy and a similar safety profile to the originator drug rituximab in treatment-naïve patients with CD20-positive B-cell lymphoma and diffuse large B-cell lymphoma (DLBCL).
CStone Pharmaceuticals Announces Preliminary Results from Phase 1b Clinical Trial of PD-L1 Antibody Combined with Cisplatin and Fluorouracil in Patients with Esophageal Squamous Cell Carcinoma (ESCC)
CStone Pharmaceuticals Announces Preliminary Results from Phase 1b Clinical Trial of PD-L1 Antibody CS1001 in Combination with Cisplatin and Fluorouracil (CF) for the Treatment of Patients with Esophageal Squamous Cell Carcinoma (ESCC)
CS1001 is a PD-L1 antibody
Phase 1b study in patients with advanced esophageal squamous cell carcinoma who have not previously received systemic anticancer therapy for locally advanced or metastatic disease
Among 18 patients with advanced esophageal squamous cell carcinoma who received CS1001 in combination with CF chemotherapy, 14 (77.8%) achieved partial response (PR). The objective response rate (ORR) was 77.8% (14 cases), and the disease control rate (DCR) reached 88.9%.
Abemaciclib in Combination with Aromatase Inhibitors Demonstrates a Statistically Significant Prolongation of Progression-Free Survival in Chinese Postmenopausal Patients with HR+/HER2- Breast Cancer
Lilly Announces That Abemaciclib Combined with Aromatase Inhibitors Demonstrated a Statistically Significant Improvement in Progression-Free Survival in Postmenopausal Women with HR+/HER2- Advanced Breast Cancer, Primarily Chinese Patients
Abemaciclib is a CDK4/6 inhibitor
MONARCH plus is a randomized, double-blind, Phase 3 clinical study conducted in postmenopausal women with HR+/HER2- advanced breast cancer, predominantly comprising Chinese patients.
The study met its primary endpoint, demonstrating a statistically significant prolongation of progression-free survival (PFS) with abemaciclib in combination with an aromatase inhibitor (anastrozole or letrozole). Furthermore, PFS was also significantly extended in women with disease progression following endocrine therapy who received abemaciclib in combination with fulvestrant.
Mmonarch Study Reports PFS Results
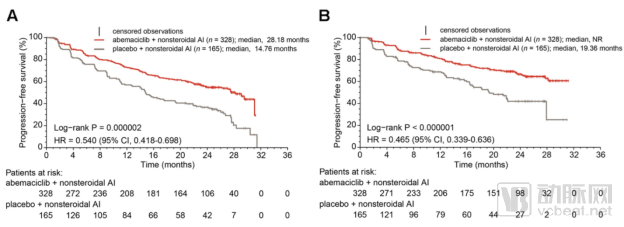
Data Source:nature
❖Roche Announces EvaluationPhase III FeDeriCa Study of Subcutaneous Fixed-Dose Combination of Perjeta and Herceptin in HER2-Positive Early Breast Cancer Meets Primary Endpoint: Efficacy and Safety Non-Inferior or Comparable to Standard Intravenous Perjeta and Herceptin Plus Chemotherapy Regimen
❖ AstraZeneca Announces, United StatesFDA Grants Fast Track Designation to Its SGLT2 Inhibitor Dapagliflozin (Brand Name: Farxiga) for the Treatment of Adult Patients with Heart Failure
❖ Roche announced that the company’s developedCD20 Antibody Gazyva Receives FDA Breakthrough Therapy Designation for the Treatment of Lupus Nephritis. This Marks Roche’s 27th Breakthrough Therapy Designation
❖Shanghai Chuangnuo's Domestic Marketing Application for Erlotinib Hydrochloride Tablets ((CYHS1790011) received approval from the National Medical Products Administration (NMPA), becoming the first domestic manufacturer to produce a generic version of this drug. The application was classified as Category 6 for registration. In September 2017, it was included in the priority review program by the Center for Drug Evaluation (CDE) as a “drug production application filed one year before patent expiration,” categorizing it as a generic drug encouraged for development by the state.