China's Innovative Drugs Enter a Golden Era: 'Made in China' Contends for 'Global Firsts' | Series on Healthcare Innovation
This year marks the 70th anniversary of the founding of the People's Republic of China. On the eve of National Day, VCBeat has specially curated a “Series on Medical Innovation,” aiming to showcase the achievements made by the healthcare industry under the national initiative for independent innovation, through our coverage and analysis of new technologies and products across various medical sectors.
The past five years have witnessed the vigorous development of innovative drugs in China. In 2018, the Center for Drug Evaluation (CDE) approved nine domestically produced Class 1 innovative drugs, setting a new historical record. Among the innovative drugs approved in recent years, there are many representative products, such as three domestic PD-1 monoclonal antibodies that keep pace with global advancements, Roxadustat—the first new drug launched globally in China—and Benvitimod, a First-in-Class drug independently developed in China. These achievements are the result of the relentless efforts made by domestic innovative pharmaceutical enterprises.
In addition to the efforts of the innovative drug industry itself, the new environment for Chinese innovative drugs, shaped by policy and capital, has played a key supportive role. At the national policy level, state authorities have continuously issued documents to lay the policy foundation for the development of the innovative drug industry. In terms of the capital market, the successive openings of the Hong Kong Stock Exchange and the STAR Market have provided domestic innovative drug companies with more sources of funding. Regarding international alignment, China’s accession to the ICH has directly integrated China’s innovative drug industry into the global landscape, bringing Chinese-made innovative drugs to a broader stage.
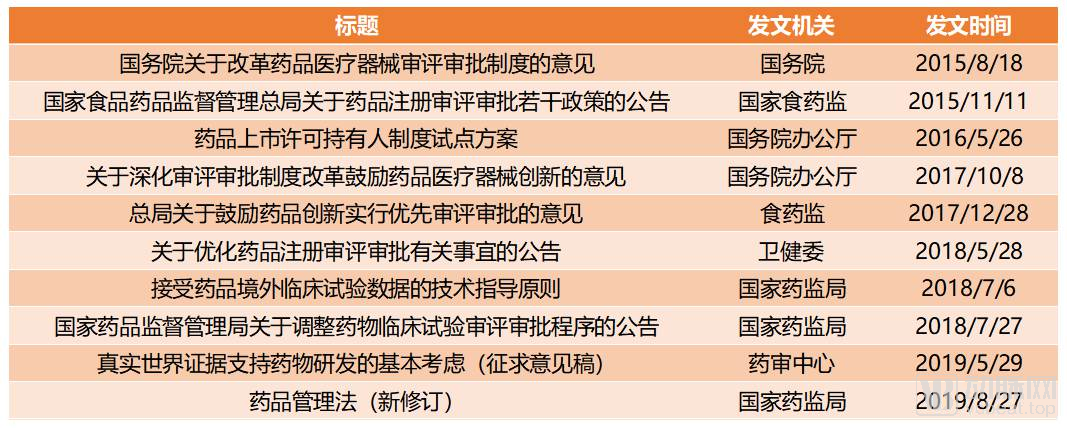
Major National Policies on Promoting the Development of Innovative Drugs Since Document No. 44
In August 2015, the State Council issued Document No. 44 [2015], titled “Opinions on Reforming the Review and Approval System for Drugs and Medical Devices” (hereinafter referred to as “Document No. 44”), officially ushering in a new era of regulatory reform for innovative drugs in China. Following Document No. 44, Document No. 42 [2017] issued by the General Office of the CPC Central Committee and the General Office of the State Council, titled “Opinions on Deepening the Reform of the Review and Approval System to Encourage Innovation in Drugs and Medical Devices” (hereinafter referred to as “Document No. 42”), further emphasized the contents outlined in Document No. 44, with particular focus on key areas such as “promoting drug innovation and the development of generic drugs,” “reforming clinical trial management,” “accelerating market approval review and processes,” and “strengthening lifecycle management of drugs and medical devices.”
Looking back, Document No. 44 and Document No. 42 have played a overarching role in the innovative drug-related policies in recent years. The key points mentioned in these two documents, after being validated over several years, were ultimately incorporated into the latest revision of the "Drug Administration Law" in 2019, becoming formal laws and regulations.
In addition to conceptually emphasizing the encouragement of innovation in the pharmaceutical industry, three key initiatives in recent healthcare policy reforms—accelerated clinical trial approvals, expedited drug review and approval processes, and the Marketing Authorization Holder (MAH) system—can be regarded as the “three carriages” through which relevant authorities have taken concrete actions to drive industry development.
1. Accelerated Clinical Approval: From 18 Months to 2 Months
Prior to the introduction of the implied permission system, the clinical trial approval process—from initial application to final issuance of the approval document—typically took approximately 18 months. In contrast, this timeframe is five days in Australia; one month in the United States and South Korea; one to two months in Singapore; three months in the European Union; and three to four months in India and Russia.
The more significant issue is that, within this 18-month period, the actual review time amounted to only about two months, with the remainder spent on various unnecessary procedures. Prior to submission to the Center for Drug Evaluation (CDE) for approval, applications must first be reviewed by provincial medical products administrations. After submission to the CDE, applicants face a queueing process that can last from several months to as long as a year. Furthermore, clinical trials cannot commence immediately after evaluation; the CDE’s evaluation report must be transferred to the Department of Drug Registration for review. Following this review, the approval document must be routed back to the provincial administration for internal processing. This multi-stage transfer process results in additional delays of several months before the documents finally reach the enterprise. Consequently, the approval process for clinical trials has long been a persistent pain point for innovative pharmaceutical companies in China.
Following the issuance of Document No. 44, the China Food and Drug Administration (CFDA), now the National Medical Products Administration (NMPA), responded actively and promptly issued the Announcement of the China Food and Drug Administration on Several Policies for Drug Registration Review and Approval at the end of 2015. The Announcement changed the regulatory requirement for bioequivalence studies of generic drugs from an approval-based system to a filing-based system, marking the initial attempt to accelerate the clinical trial approval process for generic drugs.
On July 27, 2018, the Center for Drug Evaluation (CDE) issued Document No. 50 of 2018, titled “Announcement of the National Medical Products Administration on Adjusting the Review and Approval Procedures for Drug Clinical Trials” (hereinafter referred to as “Document No. 50”). Document No. 50 explicitly states that if an applicant does not receive any negative or questioning feedback from the CDE within 60 days from the date of acceptance and payment of fees, the applicant may proceed with the clinical trial in accordance with the submitted protocol.
Not only were policy updates implemented swiftly, but the Center for Drug Evaluation (CDE) also demonstrated unwavering clarity in execution. On November 5, 2018, a new section titled “Public Notice of Implied Approval for Clinical Trials” was added to the Hot Topics column on the CDE’s official website, with eight accepted application numbers granted implied approval announced on the same day. This occurred just over three months after the release of Document No. 50, marking the formal entry of China’s clinical trial application process into the era of implied approval.
Implicit approval has drastically streamlined the previously redundant approval process at the institutional level. The Center for Drug Evaluation completes formal review within five days of receiving application materials. A notice of acceptance is issued if the materials meet requirements or comply with them after correction as prescribed. Coupled with the 60-day implicit approval period following acceptance, a clinical trial application with complete documentation requires only 65 days from submission to initiation, accelerating the previous approval timeline by one and a half years. Since the launch of the public registry for implicit approvals, 1,040 clinical trials have been approved through this mechanism. The acceleration of clinical trial approvals has yielded significant results.
2. Accelerated Marketing Approval: Clearing the Backlog of Pending Approvals and Establishing a Priority Review Mechanism
In September 2015, the number of drug registration applications awaiting review and approval peaked at nearly 22,000. State Council Document No. 44 was issued at this opportune moment, targeting the apex of the continuously rising curve. Driven by these policies, the China Food and Drug Administration (CFDA) released more specific policy guidelines at the end of the year, requiring separate review queues for eight major categories of drugs to accelerate the review and approval process.
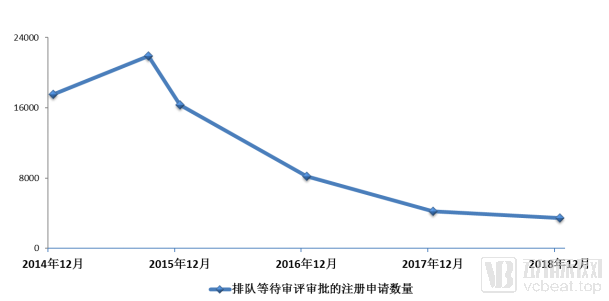
Trends in the Number of Registration Applications Awaiting Review and Approval
Under the new policy, the Center for Drug Evaluation began to accelerate the clearance of a large backlog of drug applications. By December 2017, the number of pending reviews had plummeted from 22,000 to 4,000, preliminarily achieving the work targets outlined by the State Council in Document No. 44.
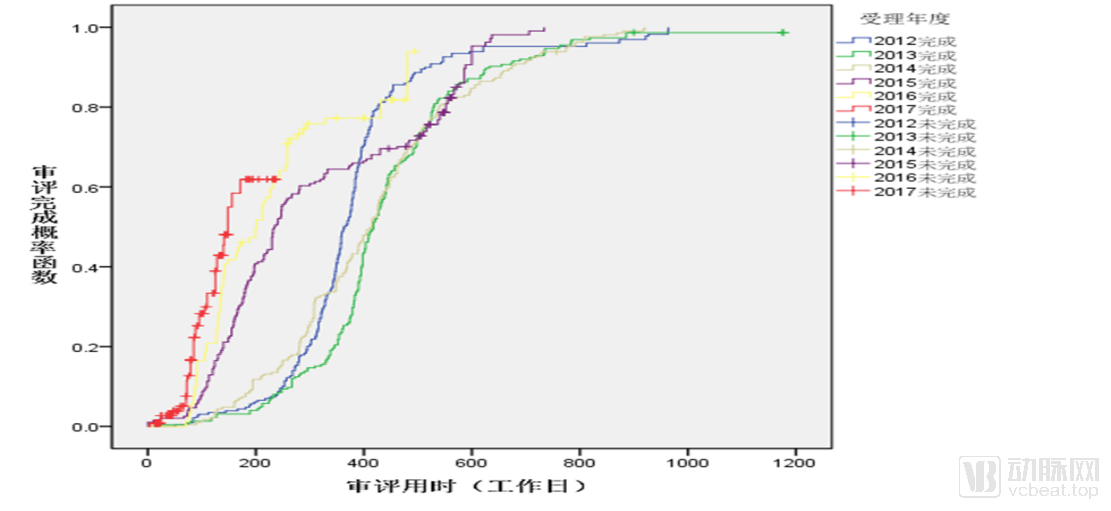
Annual Changes in NDA Review Timelines
Meanwhile, the average approval time for drugs has been significantly reduced. The median review time for New Drug Applications (NDAs) in 2017 was shortened to around 180 days, whereas this figure was over 400 days in 2013.
In December 2017, the China Food and Drug Administration (CFDA) issued the “Opinions of the General Administration on Encouraging Drug Innovation and Implementing Priority Review and Approval,” which further clarified the implementation approach for overall drug priority review. This included key aspects of the implementation process such as the scope of priority review and approval, procedures, and operational requirements, and established clear timelines for the review process to ensure that priority review and approval work proceeds efficiently and in an orderly manner.
Accelerated approval of clinical trials and expedited drug review and approval processes have jointly streamlined the entire drug approval lifecycle, freeing innovative drugs from the burden of sluggish regulatory efficiency. This development serves as a significant boost to the innovative drug industry, which is characterized by substantial R&D investments and prolonged development timelines. Expedited review also enables innovative drug companies to conduct more international multi-center clinical trials. The historical time lag between domestic and international markets, previously caused by slow domestic approvals, has now been eliminated.
3. Marketing Authorization Holder (MAH) System: Clarifying the Responsible Entity and Encouraging Technological Innovation
In the 2015 revision of the Drug Administration Law, the drug registration system still combined marketing authorization with manufacturing licensing. Specifically, drug approval numbers were issued only to manufacturers holding a Drug Manufacturing License. Enterprises and institutions lacking production capabilities were fundamentally ineligible for drug approval numbers, preventing technological innovation from being directly linked to final market returns and thereby dampening the enthusiasm of basic researchers.
On May 26, 2016, the General Office of the State Council issued the Pilot Program for the Marketing Authorization Holder System for Drugs. The Program requires that pilot implementation of the marketing authorization holder system be carried out in ten provinces (municipalities), and provides comprehensive provisions on the obligations and liabilities of marketing authorization holders, the obligations and liabilities of contracted manufacturing enterprises, and regulatory measures.
The “Plan” states that drug marketing authorization holders may “entrust drug manufacturers with production” or “entrust drug distributors with distribution.” Marketing authorization holders are only required to supervise the production and distribution processes and ensure drug traceability. This system has thoroughly broken the previously rigid integrated model of research, production, and sales in China, enabling enterprises across various segments to form flexible partnerships, fulfill their respective responsibilities, and enhance the efficiency of pharmaceutical innovation.
The Marketing Authorization Holder (MAH) system represents a key institutional framework for the development of China’s pharmaceutical industry. First, it allows basic researchers, as MAHs, to reap the ultimate market benefits derived from technological innovations, thereby incentivizing active R&D efforts. Second, it clearly identifies the party liable for personal injuries caused by marketed drugs, prompting MAHs to continuously and proactively monitor the safety and efficacy of their products. Third, it grants MAHs the authority to outsource production, helping to revitalize idle production lines, reduce redundant manufacturing, and improve capacity utilization in China’s pharmaceutical manufacturing sector.
The implementation of the Marketing Authorization Holder (MAH) system has also given rise to specialized roles in China’s pharmaceutical industry chain, such as CROs (Contract Research Organizations), CMOs (Contract Manufacturing Organizations), and CDMOs (Contract Development and Manufacturing Organizations). In the drug production process, specialized R&D entities can leverage the capabilities of CXO companies for research, development, manufacturing, and market launch, while retaining control over their core competencies. As the Marketing Authorization Holders, they ultimately reap the benefits.
On June 1, 2017, the International Council for Harmonisation of Technical Requirements for Pharmaceuticals for Human Use (ICH) approved the application of the China Food and Drug Administration (CFDA), making CFDA an official member of ICH. Just one year later, at the first ICH meeting in 2018, the National Medical Products Administration (NMPA) was elected as a member of the ICH Management Committee. Joining ICH represents a major trend in the development of China’s pharmaceutical industry, marks a significant turning point in domestic pharmaceutical regulation, and signifies that China’s drug regulatory system has gained international recognition.
Following China’s accession to the ICH, the efficiency of introducing imported originator drugs into the country has significantly improved. In 2018, the Center for Drug Evaluation (CDE) approved 106 new drugs, including 67 imported originator drugs.
On July 6, 2018, the National Medical Products Administration (NMPA) issued the Technical Guidelines for Accepting Overseas Clinical Trial Data for Drugs, allowing overseas clinical trial data to be included in drug registration applications in China. These guidelines laid a solid policy foundation for the subsequent introduction of new foreign drugs into the Chinese market.
In October 2018, the National Medical Products Administration (NMPA) and the National Health Commission jointly issued the “Announcement on Matters Concerning the Review and Approval of Overseas New Drugs in Urgent Clinical Need.” Given the limited initiative demonstrated by foreign pharmaceutical companies, the NMPA adopted a proactive approach by inviting these novel and specialty drugs to enter the Chinese market.
Subsequently, the Center for Drug Evaluation (CDE) successively released two batches of overseas new drugs urgently needed for clinical use, with 40 drugs in the first batch and 26 in the second. For these clinically urgent drugs, the National Medical Products Administration (NMPA) established a dedicated review channel. During the review process, clinical trial data conducted domestically are not required; instead, only research data obtained overseas and supporting materials demonstrating no ethnic differences need to be submitted. The Center for Drug Evaluation has also made specific time commitments, pledging to complete the technical review within six months after acceptance.
The benefits of joining the ICH have gradually become apparent. Domestic standards are now aligned with international benchmarks, facilitating mutual recognition of clinical data and harmonization of manufacturing standards. Innovative foreign drugs can enter the Chinese market more conveniently following mutual acceptance of clinical data, while new domestic drug products will be approved in accordance with international standards, thereby supporting their global expansion.
However, China’s pharmaceutical industry must also confront the challenges posed by ICH standards in the Chinese market. For a long time, China’s pharmaceutical sector has been plagued by extensive low-level duplication and insufficient innovation. With China’s accession to the ICH, its pharmaceutical industry has been compelled to compete within the global landscape. The National Medical Products Administration (NMPA) has successively invited two batches of overseas new drugs for accelerated approval and market entry, further accelerating the internationalization of China’s domestic pharmaceutical market. In the face of global competition, Chinese innovative drug companies have no choice but to target the global market and develop novel pharmaceutical products for worldwide adoption if they are to secure their future viability.
As China’s pharmaceutical market becomes increasingly internationalized, domestically developed innovative drugs are meeting national expectations and gradually gaining prominence in the global arena. As early as 2012, China’s first-in-class novel drug benvitimod licensed its overseas rights to GSK for up to $230 million. In September 2015, Jiangsu Hengrui Medicine licensed the overseas rights of its drug camrelizumab to U.S.-based Incyte Corporation for up to $800 million. While the impact of this deal was still being felt, Innovent Biologics entered into a global development and collaboration agreement with multinational pharmaceutical company Eli Lilly for three bispecific antibodies in tumor immunotherapy, with total milestone payments exceeding $1 billion.
On the path of Chinese innovative drugs going global, the U.S. FDA has demonstrated an accommodating stance. Richard Pazdur, Director of the Office of Hematology and Oncology Excellence at the FDA, stated at the American Association for Cancer Research (AACR) Annual Meeting that the FDA is willing to accept marketing applications based solely on clinical data from China, provided the quality of such data is sufficiently robust. Furthermore, the FDA welcomes the market entry of lower-cost PD-1/PD-L1 inhibitors in the United States.
International media and consulting firms have also begun to focus on China’s rapidly growing innovative drug industry, with specialized media platforms such as Chinabio Today emerging to cover the Chinese biopharmaceutical market. McKinsey, a globally renowned consulting firm, is closely monitoring China’s biopharmaceutical sector and has published insights suggesting that the industry is poised for transformation. The Economist has published an article titled “China’s Pharmaceutical Industry Is Growing,” which cites BeiGene as an example and describes China’s pharmaceutical industry as a rising star.
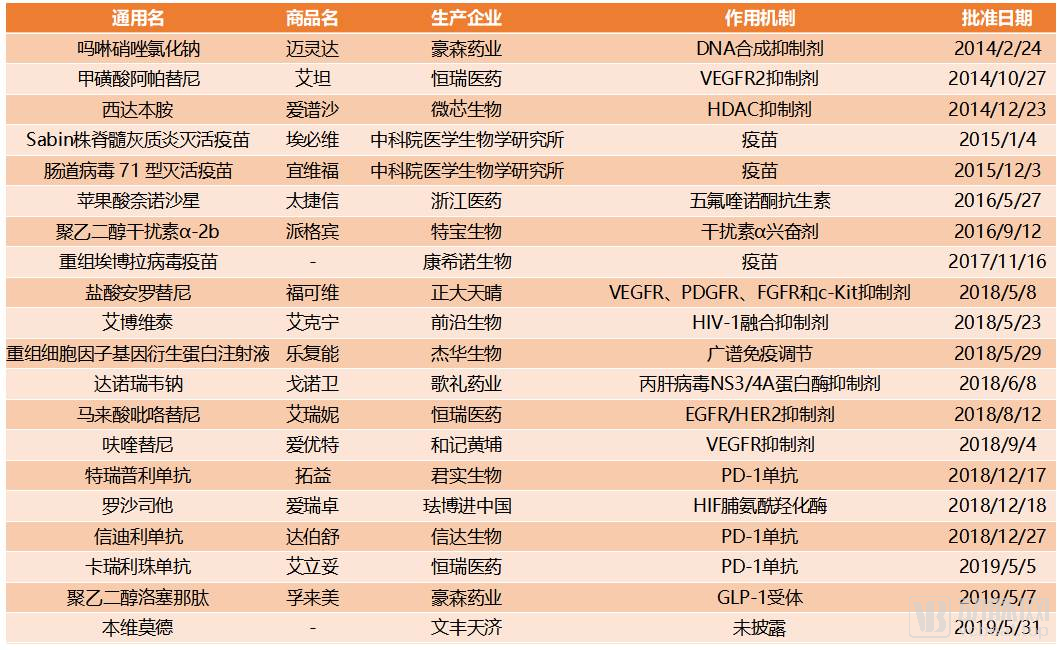
Domestic Class 1 Innovative Drugs Approved by the NMPA Since 2014
The past five years have witnessed accelerated development in China’s innovative drug industry. Notably, in 2018, the Center for Drug Evaluation (CDE) approved nine domestically produced innovative drugs for market entry, setting a historical record. While national policies played a supportive role in these approvals, the more critical factor has been the persistent commitment to innovative R&D by Chinese pharmaceutical companies. These domestic enterprises have emerged as a new force in the global pharmaceutical industry, propelling China’s innovative drug sector from a position of following to one of parity, and are poised to further strengthen their capabilities to join the ranks of industry leaders.
1. Junshi Biosciences and Innovent Biologics: The Two Leading Domestic PD-1 Players
On June 15, 2018, the China Food and Drug Administration (CFDA) approved the market launch of Bristol-Myers Squibb’s Opdivo (nivolumab), making it the first PD-1 monoclonal antibody approved for marketing in China. One month later, on July 25, the CFDA also approved the market launch of Merck & Co.’s Keytruda (pembrolizumab), formally introducing tumor immunotherapy into clinical practice in China.
After Opdivo and Keytruda were launched in the Chinese market, their overall prices decreased compared to those in foreign markets. The annual treatment cost for patients with non-small cell lung cancer using Opdivo is approximately RMB 360,000, while for melanoma patients using Keytruda, it is around RMB 190,000 per year. However, such prices still impose a significant financial burden on domestic patients. Consequently, domestically produced PD-1 monoclonal antibodies, which offer high quality at affordable prices, have become highly anticipated by patients.
The National Medical Products Administration (NMPA) did not keep patients waiting for long. On December 17, 2018, Junshi Biosciences’ Tuoyi (toripalimab) was launched, becoming the first domestically produced PD-1 monoclonal antibody to receive market approval. Just ten days later, Innovent Biologics’ Tyvyt (sintilimab) also gained approval, officially ushering in the era of domestically produced tumor immunotherapies in China. Under the patient assistance program, the annual treatment cost for malignant melanoma patients using Tuoyi is only RMB 93,600, less than half the price of Keytruda. For patients with non-Hodgkin lymphoma, the annual treatment cost with Tyvyt is just RMB 170,000.
In its 2019 interim report, Junshi Biosciences revealed that Tuoyi (toripalimab) generated total sales of RMB 303 million following its launch in late February. Innovent Biologics’ Tyvyt (sintilimab), launched in mid-March, achieved even stronger results, recording sales of RMB 332 million in just over three months. In China’s rapidly growing PD-1 market, domestically produced PD-1 inhibitors have gradually become competitive with imported products. Now, Hengrui Medicine has entered the fray with its PD-1 monoclonal antibody AiRuiKa (camrelizumab), marking the beginning of the second phase of competition focused on indications and combination therapies. We look forward to seeing more surprises from domestic PD-1 monoclonal antibodies in their future development.
2. Hengrui Medicine: A Chinese Giant Focused on Innovative Drugs
When it comes to Hengrui Medicine, many people think of a traditional domestic pharmaceutical giant whose main products include oncology drugs, anesthetics, contrast agents, and specialized infusion solutions. In fact, however, innovation has been ingrained in Hengrui Medicine’s DNA since its inception.
Imrecoxib is a Class 1 new drug approved in China in 2011 for the relief of pain symptoms associated with osteoarthritis. Hengrui Medicine initiated related research as early as 1999, entered Phase I clinical trials in 2002, and was included in the National “863 Program” by the Ministry of Science and Technology. Although subsequent sales performance was modest, imrecoxib holds symbolic significance for Hengrui Medicine that far exceeds its actual economic value. The approval of imrecoxib underscores Hengrui Medicine’s R&D capabilities as an innovative pharmaceutical company.
In November 2014, Apatinib Mesylate Tablets were approved for market launch, officially elevating Hengrui Medicine’s R&D capabilities to an international level. Apatinib is the world’s first small-molecule anti-angiogenic targeted therapy proven safe and effective in advanced gastric cancer, and it remains the most efficacious monotherapy after failure of standard treatment for advanced gastric cancer. In June of that year, the clinical study of this drug was selected as a plenary presentation by the American Society of Clinical Oncology (ASCO), marking the first time that research on an innovative Chinese drug was presented as a plenary session at a top-tier global academic conference and the first time such research was honored as an outstanding study at the annual meeting.
According to Hengrui Medicine’s 2018 annual report, the annual sales of apatinib reached RMB 1.741 billion, making it the company’s top-selling product and accounting for 10% of its total revenue. Meanwhile, Hengrui Medicine is actively expanding the indications for apatinib, having successfully established its positioning in lung cancer, breast cancer, and combination immunotherapy, while vigorously advancing clinical research.
Following Apatinib, Jiangsu Hengrui Medicine saw the successive market launches of Pyrotinib Maleate and Camrelizumab. The former is a Class 1.1 innovative drug indicated for breast cancer, while the latter is the third domestically produced PD-1 monoclonal antibody to receive approval. Beyond its marketed products, Hengrui Medicine’s R&D pipeline includes nearly 30 candidates that have entered clinical trials. As the “leading listed pharmaceutical company in China,” Hengrui is making significant strides in innovative research and development.
3. Betta Pharmaceuticals: Ushering in the Era of Domestically Produced EGFR-Targeted Drugs
In 2010, the imported targeted drug gefitinib (Iressa), an EGFR tyrosine kinase inhibitor (TKI) from AstraZeneca, was launched in China, but its exorbitant price deterred most patients.
Betta Pharmaceuticals’ icotinib hydrochloride is China’s first independently developed small-molecule targeted anticancer drug. The Phase III ICOGEN clinical trial of icotinib was the world’s first registrational Phase III trial to use a head-to-head comparison between kinase inhibitors, with AstraZeneca’s gefitinib serving as the comparator.
At that time, nearly all of China’s leading oncology hospitals participated in this trial. The Principal Investigator (PI) was Academician Sun Yan from the Cancer Hospital, Chinese Academy of Medical Sciences; the Co-Principal Investigator (Co-PI) was Professor Zhang Li from Sun Yat-sen University Cancer Center; and Professor Shi Yuankai from the Cancer Hospital, Chinese Academy of Medical Sciences, served as Chairman of the Expert Committee. A total of 27 hospitals across nine cities—including Beijing, Shanghai, Guangzhou, Nanjing, Hangzhou, Changsha, Xi’an, Chongqing, and Changchun—participated in this multicenter trial. The trial enrolled a total of 400 patients with advanced non-small cell lung cancer (NSCLC) and was completed on November 13, 2009, after ten months. After excluding one ineligible patient, the final analysis included 399 participants: 200 in the icotinib group and 199 in the gefitinib group.
In May 2010, the unblinding results of the ICOGEN trial were announced in Hangzhou, a moment that filled Beta Pharma’s R&D team and the clinical trial institutions with intense anxiety. Had the results been suboptimal, it would have not only meant that nearly a decade of Beta Pharma’s research efforts had come to naught, but also that the substantial investments made by investors, the company, and the state had been wasted.
Excitingly, the final unblinded results revealed that icotinib demonstrated favorable clinical efficacy. In second- and third-line treatment of patients with advanced non-small cell lung cancer (NSCLC), icotinib showed comparable efficacy to gefitinib but with fewer adverse reactions. The median progression-free survival (PFS) was 4.6 months for icotinib versus 3.4 months for gefitinib, with no statistically significant difference. The overall incidence of adverse events was 60.5% for icotinib compared to 70.4% for gefitinib, indicating a better safety profile and improved patient tolerability for icotinib.
The launch of icotinib achieved several firsts: it was the subject of the world’s first registration Phase III clinical trial for a tyrosine kinase inhibitor (TKI) using an active-comparator design; it was Asia’s first TKI-targeted anticancer drug; it was China’s first small-molecule anticancer drug with independent intellectual property rights; and it marked the first Phase III clinical trial in China to employ a head-to-head, double-blind, active-controlled design against an imported patented drug. Icotinib has garnered significant attention from the oncology community both in China and worldwide, as it not only achieved “me-too” status in terms of efficacy but also demonstrated “me-better” performance regarding adverse reactions. Leveraging this success, Beta Pharma has risen to the forefront of domestic innovative drug research and development, emerging as a new force in China’s homegrown innovative pharmaceutical sector.
4. Tasly Biopharmaceuticals: Creator of China’s Third-Generation Thrombolytic Drug
Puyouke, manufactured by Tasly Biopharmaceuticals, is the world’s only marketed pro-urokinase product expressed in Chinese hamster ovary (CHO) cells. It is produced through genetic engineering techniques using CHO cell expression systems and is indicated for thrombolytic therapy in acute ST-segment elevation myocardial infarction (STEMI), classifying it as a third-generation thrombolytic agent. Puyouke exerts its selective thrombolytic effect primarily by activating plasminogen on the surface of fibrin and has been included in multiple clinical practice guidelines for cardiovascular diseases.
Currently, the mainstream thrombolytic agents in the market are alteplase and urokinase, which are primarily used in hospitals lacking the capability to perform percutaneous coronary intervention (PCI). As a representative third-generation thrombolytic agent, Puyouke exhibits fibrin specificity and lacks antigenicity and allergenic potential. Data from 2,088 cases in its Phase IV clinical trials demonstrated that the drug achieved a vessel recanalization rate of 85.2% in patients with acute myocardial infarction, with an incidence of drug-related intracranial hemorrhage as low as 0.19%. Furthermore, it has a half-life of 114 minutes, offering high overall cost-effectiveness.
In 2017, Puyouke was included in the National Reimbursement Drug List (NRDL) through price negotiations conducted by the Ministry of Human Resources and Social Security, with a price reduction of 11.5% and a reimbursement price of RMB 1,020 per vial. Sales of Puyouke amounted to RMB 38 million in 2016 and surged to RMB 99 million in 2017. Following its inclusion in the NRDL, Puyouke became the leading thrombolytic product, driving continued substantial sales growth in 2018. In 2018, sales of Puyouke exceeded RMB 220 million, representing a year-on-year increase of 129.58% compared to 2017.
Tasly Biologics submitted its IPO application to the Hong Kong Stock Exchange in June this year, with its 14 R&D pipelines primarily focused on cardiovascular and cerebrovascular diseases, oncology, and digestive and metabolic disorders. Among its pipeline candidates, Puyouke’s Phase III clinical trial for ischemic stroke is progressing smoothly, while its indication for acute pulmonary embolism has entered Phase II clinical trials. Tasly Biologics holds promising prospects for further developments in the future.
5. MicroCore Bio: Second Surge in Popularity After Listing on the STAR Market
On the morning of July 17, 2019, media reports suggested that Chipscreen Biosciences’ IPO might be “withdrawn.” However, that same afternoon, the China Securities Regulatory Commission (CSRC) announced the approval of Chipscreen Biosciences’ listing, swiftly debunking the rumor. Chipscreen Biosciences debuted on the STAR Market with an issue price of RMB 20.43 per share. On its listing day, August 12, the opening share price surged to RMB 125, representing a 512% increase over the issue price. This dramatic volatility instantly propelled Chipscreen Biosciences into the spotlight.
Chidamide, developed by Chipscreen Biosciences, was actually approved for market launch as early as 2014 for the treatment of peripheral T-cell lymphoma. However, due to its narrowly approved indications, it failed to attract significant attention. Chidamide is a histone deacetylase (HDAC) inhibitor. Currently, only three companies worldwide produce drugs in this class, with two based in the United States charging monthly treatment costs of RMB 280,000 and RMB 140,000, respectively. In contrast, the monthly cost of chidamide is just over RMB 20,000, amounting to only one-tenth of the price charged abroad.
Chipscreen Biosciences has been continuously advancing further research on chidamide, striving to expand its indications, and has already achieved certain progress. The indication for breast cancer has been submitted for regulatory approval and included in the priority review and approval program. Clinical trials for non-small cell lung cancer and diffuse large B-cell lymphoma are also steadily expanding.
In addition, Chipscreen Biosciences has seven other products in its pipeline, including chiglitazar sodium and xentuzumab. The majority of the funds raised from its listing on the STAR Market have been allocated to further advancing the research and development of these products. Looking ahead, Chipscreen Biosciences will continue to innovate, taking the preservation of health as its mission. It is committed to providing patients with innovative therapeutics that are safe, highly effective, and affordable.
6. FibroGen China: Roxadustat Becomes the First Class 1.1 New Drug Globally Developed and Launched in China
On December 18, 2018, roxadustat was first approved for marketing in China for the treatment of anemia caused by chronic kidney disease (CKD) in patients undergoing dialysis. On August 20, 2019, roxadustat received approval in China for its second indication globally, expanding its labeled use to include non-dialysis-dependent chronic kidney disease (NDD-CKD) anemia. The initial launch of roxadustat in China marks the first time that a First-in-Class new drug developed in China has taken a leading position worldwide.
Preclinical studies of roxadustat were primarily conducted overseas; however, its developer, FibroGen, initiated Phase I clinical trials in China as early as 2010, established FibroGen China in 2011, and built a manufacturing facility in the country to produce the drug. In late 2017, roxadustat was included in the priority review program.
On July 25, 2019, The New England Journal of Medicine (NEJM) published back-to-back online articles presenting two studies on roxadustat, an innovative drug for renal anemia, announcing the results of its two Phase III clinical trials in China; accompanied by commentaries from internationally renowned nephrology experts. This marks the first time NEJM has published Phase III clinical trial data of a new drug with mainland Chinese physicians as both first and corresponding authors, and also the first time the journal has published back-to-back clinical trials conducted by Chinese research teams.
More than 50% of patients with stage 4–5 chronic kidney disease (CKD) exhibit symptoms of anemia, and the prevalence rises to 60–90% among dialysis patients, who suffer from anemia of varying severity. For a long time, clinical management of anemia in CKD patients has relied on the combination of erythropoiesis-stimulating agents (ESAs) and iron supplements, but this approach has yielded only modest results. Roxadustat, administered as a single oral medication, offers a solution to this clinical challenge and holds the potential to revolutionize the treatment of anemia in chronic kidney disease.
7. Wenfeng Tianji: China’s First “Global Novel” New Drug in Dermatology
On May 31, 2019, benvitimod was approved for market launch, indicated for the topical treatment of mild-to-moderate stable plaque psoriasis in adults. This first-in-class novel dermatological drug, independently developed in China, finally bore significant fruit after a 20-year long march.
Topical treatments for psoriasis primarily consist of glucocorticoids and vitamin D3 derivatives; however, corticosteroid-based therapies are associated with high recurrence rates and adverse effects. Consequently, vitamin D3 agents, represented by calcipotriol, have become first-line treatments for psoriasis and are regarded as the “gold standard” among non-steroidal topical medications.
Phase III clinical trial results for benvitimod indicate that it demonstrates efficacy comparable to, or even superior to, that of calcipotriol. Benvitimod also offers significant advantages, including rapid onset of action, sustained efficacy, low relapse rates after discontinuation, and a high safety profile, with no systemic adverse reactions observed. In other words, benvitimod has the potential to replace calcipotriol as the “new standard” in the treatment of psoriasis.
Since the first order on August 26, benvitimod has received highly positive market feedback. Although the price of RMB 496 per tube is somewhat high, overall sales remain in a state of excess demand over supply. Currently, benvitimod is primarily prescribed by hospital physicians, with patients purchasing the medication at Direct-to-Patient (DTP) pharmacies. In the future, Wenfeng Tianji will also expand and strengthen its sales team to enable more patients to access this global new drug made in China at an earlier stage.
8. CanSino Biologics: The Pride of Chinese-Made Vaccines
CanSino Biologics’ Ebola vaccine, Ad5-EBOV, received approval for its new drug application from the National Medical Products Administration (NMPA) in October 2017, allowing for emergency use and inclusion in national stockpiles. Compared with competing products from multinational corporations, this vaccine demonstrates superior stability and does not require ultra-cold storage conditions.
CanSino Biologics’ Ebola vaccine employs internationally advanced replication-deficient viral vector technology and serum-free, high-density suspension culture technology. It can simultaneously elicit cellular and humoral immune responses in the human body, ensuring safety while demonstrating favorable immunogenicity. Furthermore, this vaccine has overcome the technical bottleneck associated with lyophilized formulations of viral vector vaccines. This confers particularly significant advantages for transportation and use in high-temperature regions, such as Africa.
In addition to the Ebola vaccine, CanSino Biologics has developed 14 product pipelines in development based on its five core technology platforms, covering a range of diseases including pneumonia, tuberculosis, Ebola virus disease, meningitis, diphtheria-tetanus-pertussis, and cervical cancer.
On March 28, CanSino Biologics was officially listed on the Main Board of The Stock Exchange of Hong Kong. This IPO marked CanSino Biologics’ formal entry into the capital market as the “first vaccine stock” on the Hong Kong stock exchange. Amidst frequent challenges facing the domestic vaccine industry, CanSino Biologics stands out as a beacon of hope for Chinese-made vaccines, ushering in a new chapter for China’s vaccine sector.