Rare Disease Gene Therapy Enters a New Era of Renaissance: A Comprehensive Industry Research Report
This excerpt is taken from Probe Capital’s “Industry Research on Gene Therapy for Rare Diseases” report.
Gene therapy is a therapeutic approach that introduces normal or therapeutically active genes into the human body through specific methods to correct or compensate for diseases caused by genetic defects and abnormalities. In 1972, American biologists T. Friedman and R. Robin published an article titled “Gene therapy for human genetic disease?” in the journal Science, proposing the hypothesis of using gene therapy to treat genetic disorders. This article is widely regarded as groundbreaking and forward-looking. Since then, accompanied by a series of clinical trials, the development of gene therapy has undergone four stages: “initial exploration,” “rapid expansion,” “turbulent progress,” and “resurgent prosperity.”
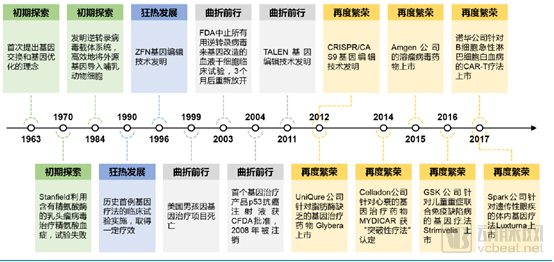
History of Gene Therapy Development (Source: Huajin Securities)
Currently, there are two basic strategies for gene therapy: the first uses integrating vectors to introduce genes into the genome of precursor cells or stem cells, allowing the inserted genes to be passed on to daughter cells during cell division; the second uses non-integrating vectors, where the introduced genes exist independently within the nuclei of host cells that lack the ability to divide or divide very slowly, thereby achieving sustained expression of the relevant genes. Based on these two therapeutic strategies, the specific procedures of gene therapy can be categorized into ex vivo and in vivo approaches.
The process of ex vivo therapy involves isolating a patient’s autologous cells, introducing therapeutic genes into these cells outside the body, and finally infusing the modified cells back into the patient. Ex vivo therapies typically employ integrating vectors to insert genes into cells with sustained proliferative capacity, such as hematopoietic stem cells. In contrast, in vivo therapies utilize non-integrating vectors and are generally administered via methods similar to those for conventional drugs, such as intravenous injection, offering greater operational feasibility.
In practical applications, gene therapy technologies are primarily categorized into two major types: viral vector-based gene delivery technologies and gene editing technologies.
Gene delivery technologies can employ strategies such as introducing normal genes to compensate for defective endogenous genes, or delivering inhibitory sequences targeting disease-causing genes (e.g., siRNA, shRNA) to reduce their transcription or translation levels.
Gene editing technologies are primarily represented by three classes: zinc finger nucleases (ZFNs), transcription activator-like effector nucleases (TALENs), and clustered regularly interspaced short palindromic repeats (CRISPR)–CRISPR-associated proteins (Cas). These technologies enable the correction, knockout, and insertion of target genes.
Currently, the core safety considerations for ex vivo and in vivo therapeutic strategies differ: ex vivo therapies focus on the risk of insertional mutagenesis during genomic integration, while in vivo therapies are concerned with excessive immune responses caused by viral infusion.
In addition, plasmid DNA and naked DNA can also be delivered via non-viral gene delivery methods, such as microinjection, gene gun, or using liposomes and nanoparticles. However, the range of applicable tissues/cells is relatively limited, transfection efficiency remains low at present, and related vectors are still under continuous development.
Genetic disorders and rare diseases represent key application areas for gene therapy, with most currently approved gene therapies focusing on these two fields. However, the high cost, the need for improved regulatory frameworks, intense competition coupled with limited indications, have sparked both widespread attention and controversy within the industry regarding gene therapy. This article will provide an in-depth look at the unique landscape of gene therapy by examining three aspects: recent advances in gene therapies for rare and genetic diseases, relevant policies, industry standards, and reimbursement systems, as well as the competitive landscape.
Current applications of gene therapy include monogenic inherited disorders, hemophilia, ophthalmic diseases, certain neurodegenerative conditions, polygenic inherited disorders with relatively well-understood pathogenic and therapeutic mechanisms, as well as cancer treatment research. This article provides a summary and analysis of drug approvals and clinical trial progress in the first category of these therapeutic areas.
Approved Gene Therapy Products
Since 2016, the European Medicines Agency (EMA) and the U.S. Food and Drug Administration (FDA) have collectively approved six gene therapy products, including two CAR-T cell therapies for B-cell hematologic malignancies and four gene therapies for severe monogenic genetic disorders. The first approved adeno-associated virus (AAV) gene therapy was Glybera, developed by the Dutch company uniQure, which received EMA approval in 2012 for the treatment of lipoprotein lipase deficiency. However, due to its limited efficacy, exorbitant pricing (averaging $1 million per treatment), and extremely rare indication (with an incidence rate of 1 in 1 million), only one patient received the therapy during its market availability. Consequently, Glybera was quietly withdrawn from the market in 2017.
In May 2016, the EMA approved GSK’s Strimvelis for marketing authorization to treat severe combined immunodeficiency (SCID) caused by adenosine deaminase (ADA) deficiency (in April 2018, GSK transferred its rare disease gene therapy pipeline, including Strimvelis, to Orchard Therapeutics in exchange for a 19.9% equity stake).
Due to severe immune system impairment, some patients with ADA-SCID die during infancy or early childhood, while others are confined to living in sterile environments for life; hence, ADA-SCID is also known as “bubble boy disease.” Prior to the emergence of this therapy, patients could only rely on hematopoietic stem cell transplantation or enzyme replacement therapy for management. Enzyme replacement therapy requires weekly injections and costs approximately $400,000 per year.
Strimvelis is an ex vivo gene therapy that uses a gamma-retroviral vector to introduce the normal ADA gene into patients’ CD34+ hematopoietic stem and progenitor cells (HSPCs). The treatment costs approximately $660,000 per dose, and in theory, a single administration provides lifelong benefit.
In December 2017, the FDA approved Luxturna, developed by Spark Therapeutics, for the treatment of Leber’s congenital amaurosis type II (LCA), a hereditary retinal disease caused by biallelic mutations in the RPE65 gene. Mutations in the RPE65 gene result in the loss of isomerase activity of its protein product, leading to a deficiency of 11-cis-retinal. This deficiency impairs the ability of photoreceptor cells to respond to light. In advanced stages, the lack of 11-cis-retinal may trigger degeneration of cone and rod cells, resulting in severe vision loss. Prior to the approval of Luxturna, there were no effective therapeutic agents available for LCA. Luxturna utilizes an adeno-associated virus serotype 2 (AAV2) vector carrying the RPE65 gene, which is administered via direct subretinal injection into the eye. The price for bilateral treatment with this therapy is approximately $850,000.
In May 2019, the FDA approved Zolgensma, an in vivo gene therapy drug developed by AveXis Inc., a subsidiary of Novartis, for the treatment of infants (under 2 years of age) with spinal muscular atrophy (SMA) caused by biallelic mutations in the SMN1 gene. The incidence of SMA is approximately 1 in 5,000 to 1 in 12,000, characterized by progressive muscle weakness and paralysis. Based on the number of SMN gene copies and protein expression levels, SMA severity is classified into four subtypes, with Type I being the most severe. Symptoms typically appear when infants are around six months old, and only 8% of affected infants survive beyond two years of age. Prior to the approval of Zolgensma, Biogen’s Spinraza was the only therapy approved for treating SMA in the United States. Unlike Zolgensma’s therapeutic approach, Spinraza is an antisense oligonucleotide (ASO) targeting the SMN gene, administered via intrathecal injection through lumbar puncture every four months. Spinraza may theoretically require lifelong administration, with a corresponding treatment cost of $750,000 for the first year and $375,000 annually thereafter. Zolgensma achieves a potential cure for SMA by delivering a functional SMN gene via an AAV9 vector through intravenous infusion. The product’s official list price is $2.125 million per one-time treatment, making it the most expensive single-treatment drug in history.
However, one month after the FDA approved Zolgensma for market release, AveXis, the developer and manufacturer of the gene therapy, voluntarily disclosed to the FDA and other regulatory authorities that certain data in an animal study had been manipulated. On August 6, 2019, the FDA initiated a comprehensive review of the accuracy of Zolgensma’s data. To date, the investigation has not identified any issues regarding the product’s safety, efficacy, or quality.
In June 2019, the European Medicines Agency (EMA) granted conditional approval to Zynteglo, a drug developed by bluebird bio, for the treatment of patients aged 12 years and older with non-β0/β0 genotype transfusion-dependent beta-thalassemia (TDT). TDT is a hereditary anemia caused by defects or mutations in the gene encoding beta-globin, leading to reduced beta-globin synthesis and an excess of alpha-globin, which forms insoluble inclusion bodies within red blood cells. This induces erythrocyte apoptosis, resulting in anemic symptoms. The global incidence of this disease is 1 in 100,000, while in the European Union it is 1 in 10,000; however, certain regions exhibit extremely high prevalence rates (Cyprus: 14%; Sardinia: 10.3%). Standard treatments for TDT include long-term blood transfusions combined with iron chelation therapy or hematopoietic stem cell transplantation.
Zynteglo is an ex vivo gene therapy that introduces a functional human βA-T87Q-globin gene into CD34+ hematopoietic stem cells (HSCs) via a lentiviral vector, followed by reinfusion, enabling patients to autonomously produce β-globin and generate sufficient hemoglobin, thereby reducing or eliminating the need for transfusion therapy. Additionally, bluebird bio is evaluating the efficacy of Zynteglo in sickle cell disease, with plans to launch it in the U.S. market in 2020. Priced at $1.8 million per one-time treatment, this therapy is the “second most expensive” drug, surpassed only by Zolgensma.
Analysis of Gene Therapy Clinical Trials
As of September 2019, there were 115 clinical trials of ex vivo gene therapy using lentiviral vectors and 190 clinical trials of in vivo gene therapy using recombinant adeno-associated virus (rAAV) vectors. A total of 32 clinical trials employed ZFN, TALEN, and CRISPR-Cas technologies as gene-editing tools, with the majority targeting cancer treatment.
In the field of rare disease and genetic disorder treatment, four studies are in vivo gene editing trials using rAAV as the vector, while five are ex vivo gene editing trials (not using lentivirus as the delivery vector for gene editing tools). Among ongoing gene therapy clinical trials, approximately 58% are Phase II studies, 33% are Phase I studies, and about 9% are Phase III clinical trials.
There are currently 55 clinical trials underway for ex vivo gene therapies using lentiviral vectors, with the majority focusing on the treatment of genetic disorders such as X-linked severe combined immunodeficiency (X-SCID), sickle cell anemia, hemophilia, mucopolysaccharidosis (MPS), and lysosomal storage diseases. A summary of selected relevant clinical trials is provided below:
Compilation of Selected Clinical Trials for Ex Vivo Gene Therapies Targeting Rare Diseases
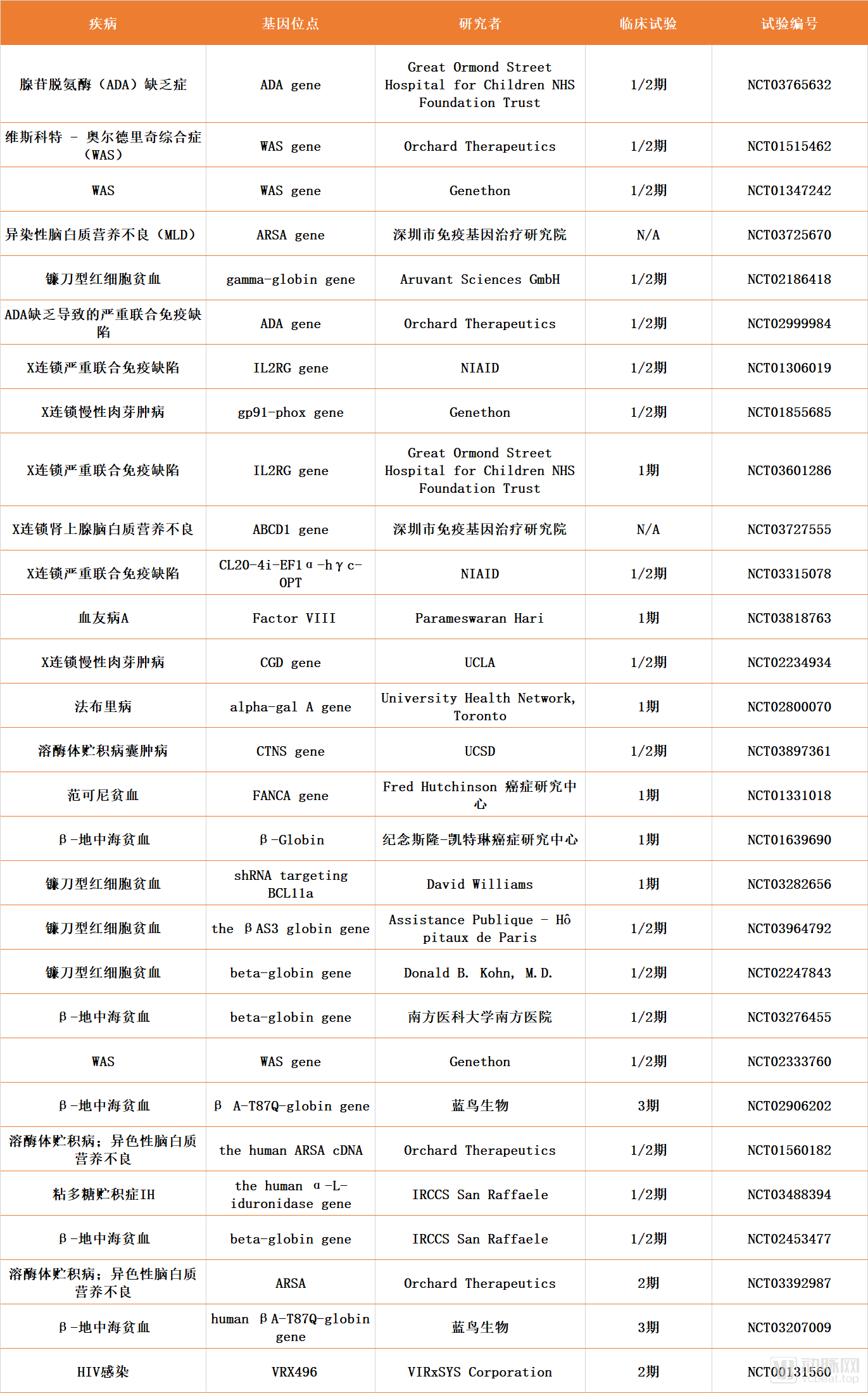
Data source: ClinicalTrials.gov, compiled by Probe Capital
Among the various serotypes, AAV2 and AAV8 have been the most extensively studied and are most frequently used in clinical trials. Due to the inherent tissue- and organ-specific tropism of different recombinant adeno-associated virus (rAAV) serotypes, the majority of current rAAV-based gene therapies focus on targeted delivery to the liver, striated muscle, and central nervous system. Various natural AAV capsid proteins can deliver transgenes to the liver, enabling this vector platform to play a therapeutic role in the treatment of hemophilia A, hemophilia B, familial hypercholesterolemia, glycogen storage disease type Ia, and Crigler-Najjar syndrome.
AAV8 and AAV9 can target multiple muscle types throughout the body for the treatment of Duchenne muscular dystrophy (DMD), as well as treat heart failure by delivering specific genes involved in certain signaling pathways and metabolism, such as SERCA2a, to the heart.
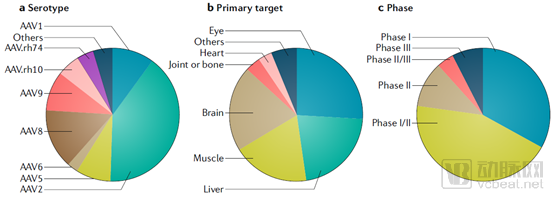
Statistics on Clinical Trials of AAV Vector-Based In Vivo Gene Therapies (as of November 2018) (Source: Nature)
As shown in the figure above, a significant proportion of recombinant adeno-associated virus (rAAV)-based gene delivery therapies in clinical development target the central nervous system, including the brain and the eye. The eye is a relatively compartmentalized organ; the presence of the blood-retinal barrier allows rAAV vectors to achieve effective gene delivery via direct subretinal injection. This approach requires a lower viral vector dose, avoids inflammatory immune responses, and mitigates the reduction in therapeutic efficacy caused by antibody neutralization or immune reactions.
Current indications for gene therapy clinical trials targeting the eye include Leber congenital amaurosis, inherited chorioretinal dystrophies, achromatopsia, and Leber hereditary optic neuropathy (LHON).
In contrast, the brain is more complex and relatively larger in volume; therefore, direct intraparenchymal injection of rAAV results in localized distribution of the vector. For neurological disorders with relatively well-defined pathological regions, such as Parkinson’s disease, rAAV administration to the nucleus capsular region represents an ideal therapeutic approach. Additionally, intrathecal injection of viral vectors into the cerebrospinal fluid space can achieve broader central nervous system distribution, albeit with relatively higher risks. AAV9 and AAVrh.10 are capable of crossing the blood-brain barrier, enabling gene delivery to neurons and glial cells. Current studies have demonstrated the efficacy of systemic rAAV therapy for central nervous system diseases, including indications such as spinal muscular atrophy (SMA), amyotrophic lateral sclerosis, and Canavan disease. The status of selected rAAV gene delivery clinical trials is summarized in the table below:
Statistics on Some rAAV Gene Delivery Clinical Trials
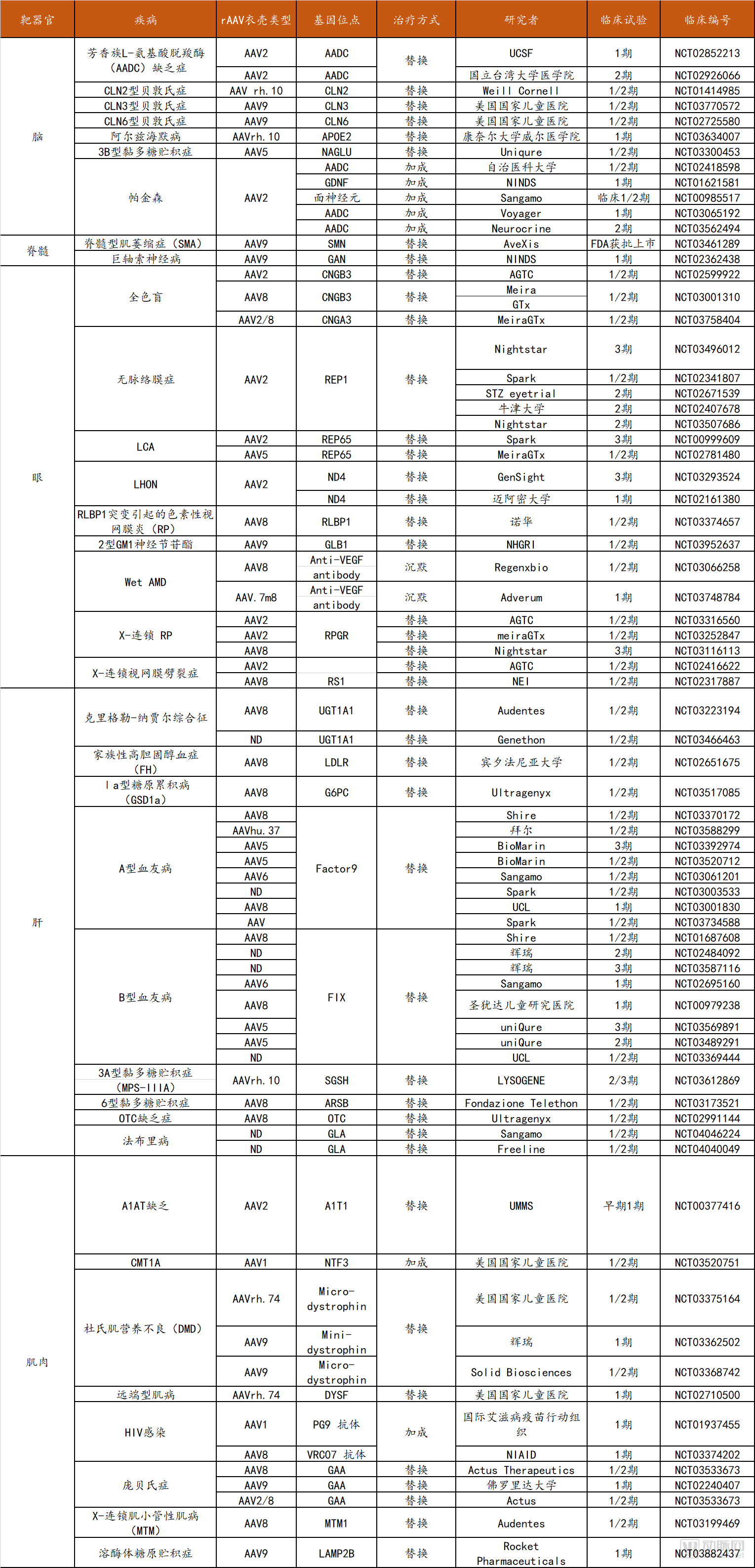
Data Source: ClinicalTrials.gov, Probes Capital
Due to the relative novelty of gene editing technologies, their mechanisms are not yet fully elucidated, and off-target effects remain incompletely controllable. Consequently, despite significant attention and extensive research within the scientific community, the clinical application of gene editing remains relatively limited due to safety concerns. To date, three types of gene editing technologies have been documented in clinical trials: ZFN (14 trials), TALEN (3 trials), and CRISPR (15 trials). The majority of ZFN clinical trials target HIV infection (8 trials, with CCR5 as the target), while most TALEN and CRISPR trials focus on the treatment of various cancers (13 trials). Furthermore, China ranks among the top countries globally in terms of the total number of clinical trials.
In the field of rare and genetic disease treatment, Sangamo Therapeutics has secured all five ZFN trials, with indications including hemophilia B, mucopolysaccharidosis (MPS), and thalassemia and sickle cell disease (conducted in collaboration with Bioverativ, a subsidiary of Sanofi). There are four CRISPR-Cas9-related trials: three involve ex vivo therapies for blood disorders, while the sole in vivo study targets Leber congenital amaurosis.
Summary of Clinical Trials on Gene Editing for Rare Diseases/Genetic Disorders
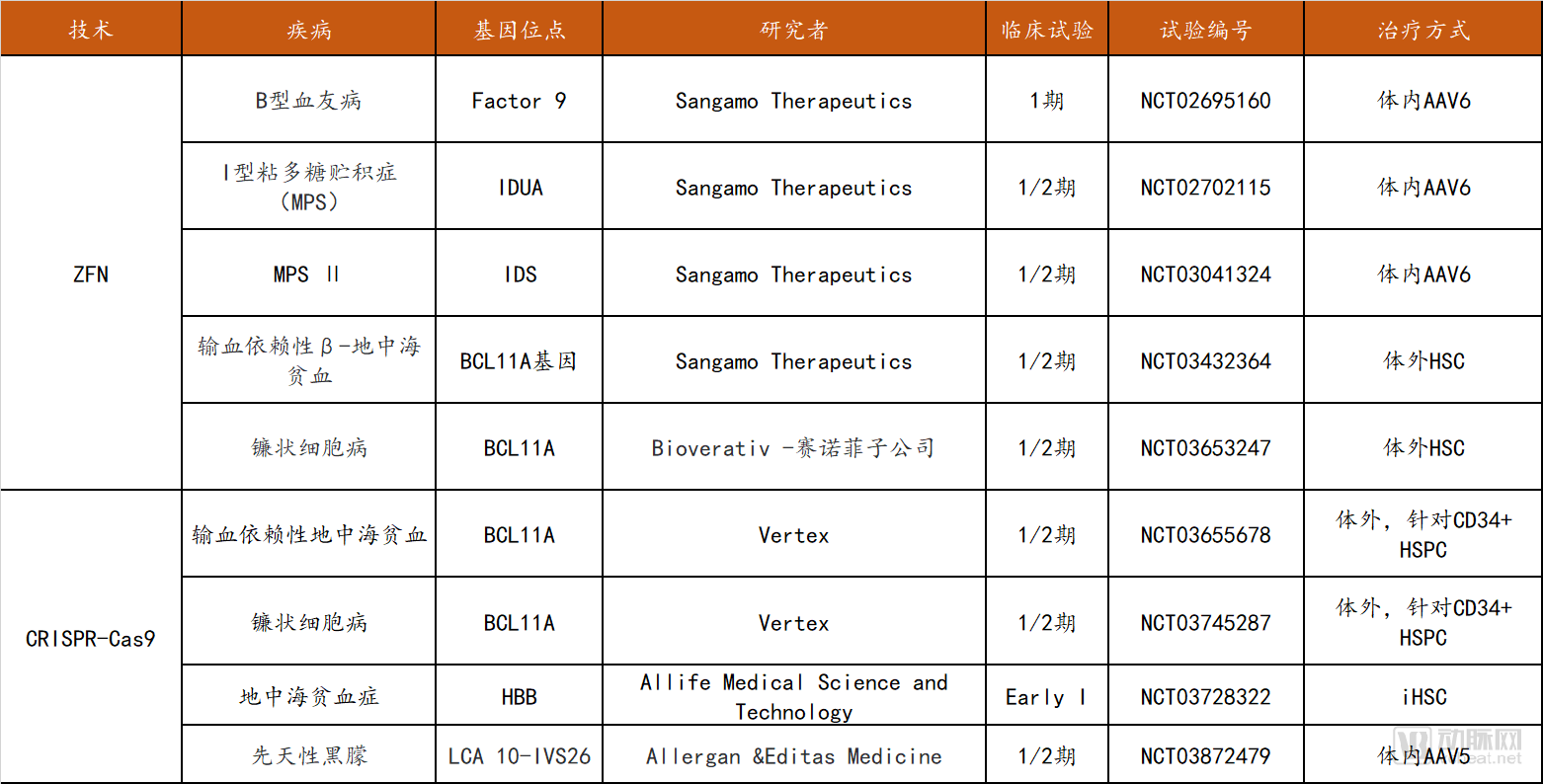
Data source: clinicaltrials.gov, Probe Capital
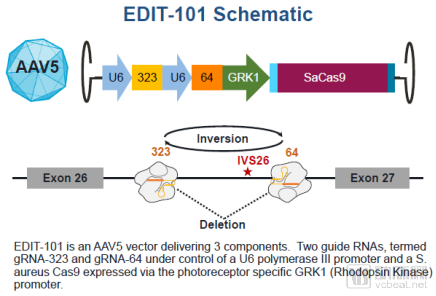
Mechanism of Action of the First In Vivo CRISPR Gene-Editing Therapy (Source: Editas Medicine Website)
Gene therapy, particularly gene editing-based approaches, as an emerging technology, is subject to varying degrees of regulation by governmental organizations across different regions worldwide. Europe and the United States have established relatively rigorous and comprehensive regulatory frameworks in this field, whereas China’s laws, regulations, and oversight mechanisms require further development. In addition, several international consortia within the industry play a significant role in standardizing the technology and its clinical applications.
Specifically regarding gene therapy for rare diseases, 70% of current gene therapies target rare conditions. In recent years, multiple countries and regions, including the United States, the European Union, Japan, and China, have introduced a series of favorable policies for rare diseases, such as tax credits, special fund grants for research, market exclusivity periods, and accelerated registration, review, and approval processes. When applying for clinical trials, rare disease therapies may appropriately reduce the number of clinical trial cases or apply for exemptions from conducting clinical trials. For instance, MB-107, a lentiviral vector gene therapy for X-linked severe combined immunodeficiency (X-SCID) developed through a collaboration between Mustang Bio and St. Jude Children’s Research Hospital, was granted Regenerative Medicine Advanced Therapy (RMAT) designation by the United States. RMAT is a review pathway specifically established for regenerative therapies, similar to the FDA’s Breakthrough Therapy designation. This designation will accelerate the development and review process of the therapy.
Following the U.S. launch of Luxturna, a therapeutic agent for congenital amaurosis, in December 2017, China’s National Medical Products Administration (NMPA) included it in the first public list of overseas drugs urgently needed for clinical use on November 1, 2018. The drug is currently undergoing expedited review and approval. This development demonstrates a significant policy shift toward supporting rare disease treatments, thereby encouraging the introduction, research and development, and manufacturing of gene therapies for rare diseases. Furthermore, given the exorbitant pricing of currently marketed gene therapy products for genetic disorders, the reimbursement framework for such therapies has become a focal point of widespread attention.
Overview of U.S. Gene Therapy Policies
The United States is a pioneer in gene therapy, with a relatively comprehensive regulatory framework. Compared to regulatory agencies in other countries, the U.S. tends to adopt a conservative and cautious approach. For instance, the European Union approved its first gene therapy drug in 2012, whereas the FDA did not approve its first gene therapy drug until 2017.
As early as 1974, the U.S. National Institutes of Health (NIH) established the Recombinant DNA Advisory Committee (RAC), whose mission expanded from overseeing research involving emerging technologies for manipulating nucleic acids to encompassing the review and discussion of human gene therapy protocols. In 1984, the FDA began regulating gene therapy products for the first time, establishing a dual regulatory framework involving both the FDA and the NIH. In 1990, the FDA started approving clinical trials for human gene therapies. However, a fatal incident in gene therapy occurred in 1999, prompting the government to strengthen oversight and regulation of gene therapy. The FDA and NIH subsequently issued multiple regulatory guidelines for gene therapy, while specific regulatory approaches have continued to evolve.
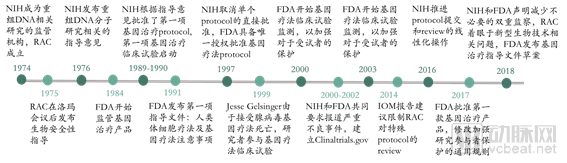
The Regulatory History of Gene Therapy in the United States (Source: NEJM, VCBeat)
In August 2018, the FDA and NIH jointly published an article in the NEJM, stating that there is insufficient evidence to suggest that gene therapy poses unique or unpredictable safety risks, and thus does not require regulatory measures different from those for other therapies. Consequently, the United States will reform its existing regulatory framework for gene therapy, gradually streamlining regulatory functions under the single agency of the FDA to encourage the development of gene therapies. In line with continuous advancements in gene therapy technology, the FDA provides guidance to companies at every stage of product development and has issued a total of 12 guidelines covering seven key areas.
FDA Regulations on Gene Therapy
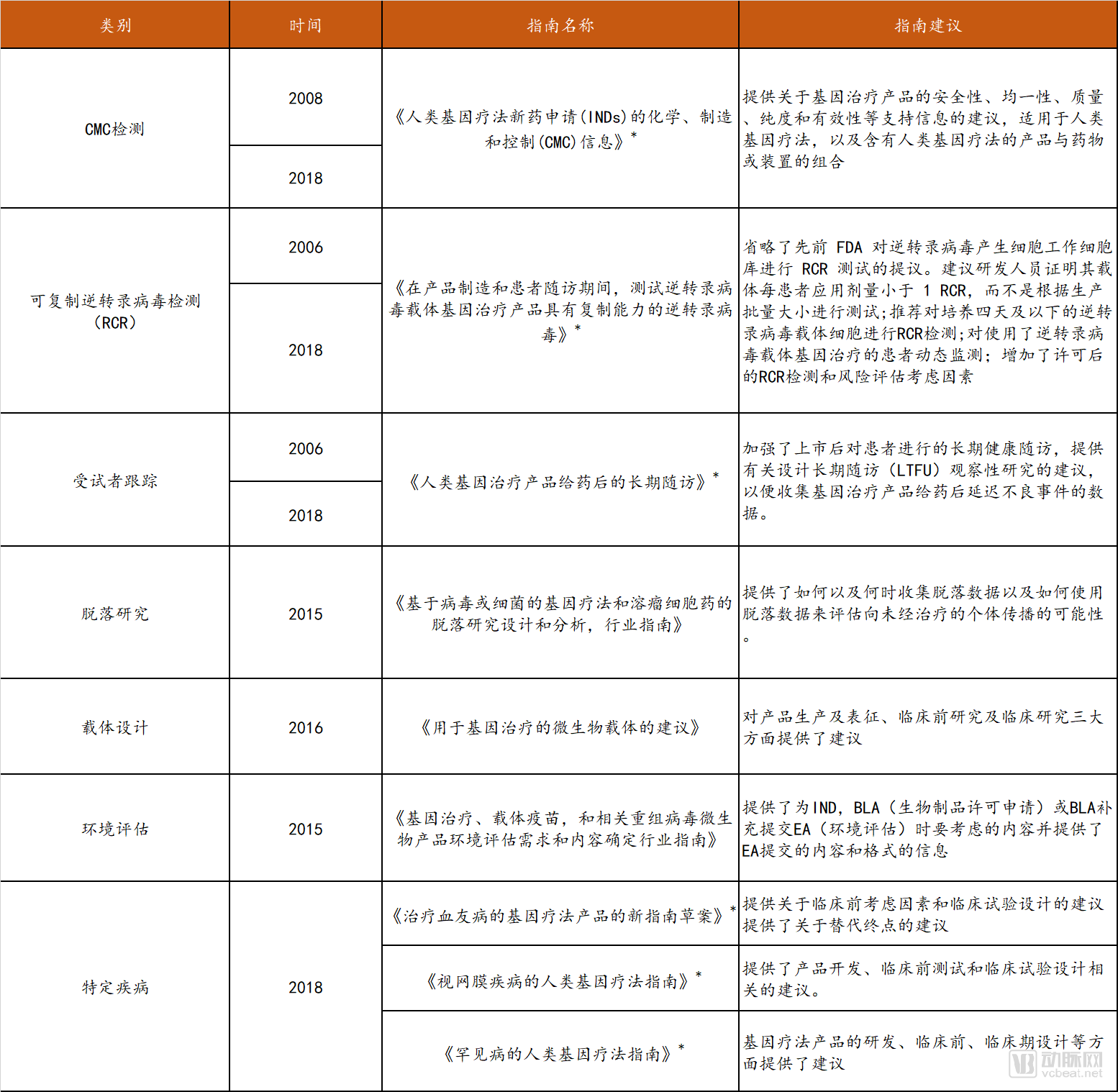
* Draft
Data source: FDA official website, compiled by Probe Capital
Taking the 2018 “Guidelines on Human Gene Therapy for Rare Diseases” (Draft) as an example, this guideline aims to assist sponsors in designing clinical development plans by providing recommendations on challenges such as limited study populations and scale, potential feasibility and safety issues, and difficulties in interpreting efficacy.
Overview of Domestic Gene Therapy Policies
In the field of gene therapy, China initiated basic research and clinical trials relatively early, conducting two clinical trials in 1991 on ex vivo retroviral therapy for hemophilia B. However, policies and regulations have long lagged behind, remaining relatively simplistic and lacking detailed provisions or specifications for specific issues arising during the research process, resulting in weak regulatory enforcement. In the early stages, domestic approval processes for such products were relatively lenient. For instance, in 2003, a gene therapy drug targeting head and neck tumors via the P53 gene, named “Gendicine,” was approved for market launch despite having only slightly over 100 cases in clinical trials and lacking standard Phase III clinical trial data. It was marketed as the “world’s first approved gene therapy drug.” Furthermore, the “world’s first gene-edited babies” incident in late 2018, which shocked the global community, severely damaged the image and interests of China’s scientific and technological community and relevant government agencies.
In this context, since 2019, relevant government agencies have been continuously strengthening regulation over the gene therapy sector:
The “Draft Second Deliberation Version of the Personality Rights Part of the Civil Code,” released in April 2019, added provisions stipulating that medical and scientific research activities involving human genes, human embryos, and related matters shall comply with laws, administrative regulations, and relevant national provisions; they must not endanger human health or violate ethical morals. The third deliberation draft of the Personality Rights Part, submitted on August 22, further added the provision that such activities “shall not harm public interests.” In May 2019, the “Regulations on the Management of Human Genetic Resources of the People’s Republic of China” were issued, making corresponding revisions and improvements to gene editing in light of the latest developments.
In 2017, the Chinese Pharmacopoeia Commission initiated a project to draft general technical requirements for gene therapy products, drawing on guidelines from the U.S. Food and Drug Administration (FDA) and the United States Pharmacopeia (USP). In June 2019, it released the “General Chapter on Human Gene Therapy Products (Draft for Public Comment),” which established foundational standards to ensure product safety and efficacy, thereby promoting the industrialization and clinical application of such technologies. Furthermore, the State Council announced that, to strengthen regulation and oversight of life sciences research and medical activities—including “gene editing”—legislative efforts would be accelerated in 2019 regarding safety management in biotechnology R&D and clinical application management of new biomedical technologies, aiming to establish a comprehensive regulatory framework covering the entire process. It is anticipated that gene therapy-related technologies will develop in China with greater rigor in the future. The current relevant laws and regulations pertaining to gene therapy in China are summarized below.
Regulations on Gene Therapy in China
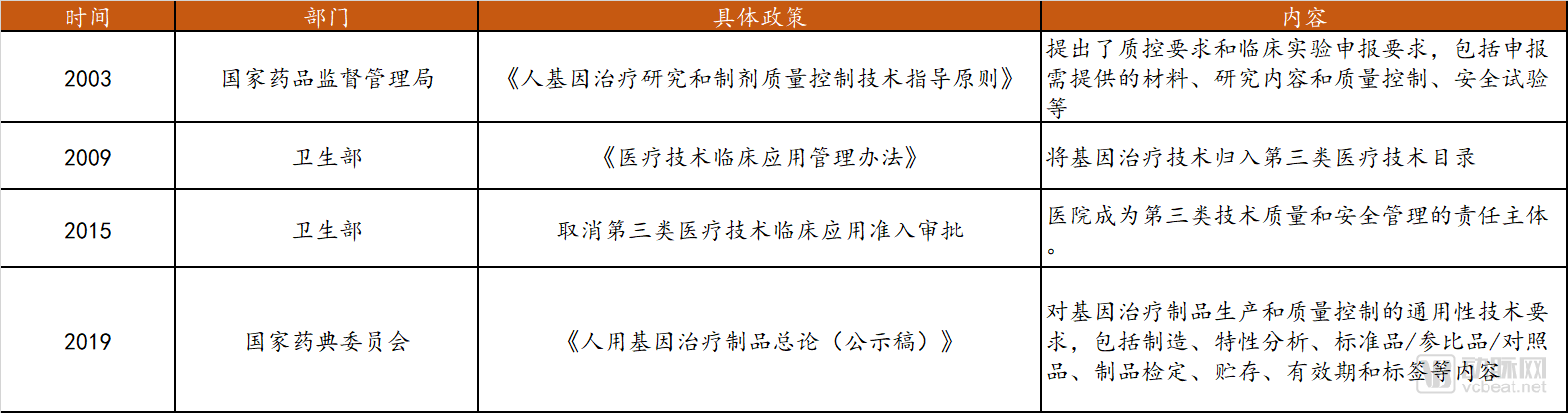
Data source: Public information, compiled by Probe Capital
Industry Standards for Gene Therapy
In addition to oversight and guidance from relevant government agencies, there are also international industry consortia addressing gene therapy as a novel treatment modality, such as the NIST Gene Editing Consortium, the International Organization for Standardization (ISO), and the Alliance for Regenerative Medicine (ARM). At the behest of scientists and related enterprises, these consortia have established an international bioethics framework for gene therapy.
ARM, an international advocacy organization for cell and gene therapy and the broader field of regenerative medicine, recently spearheaded the development of the Statement of Principles for Gene Therapy Developers. The Statement of Principles outlines five key principles for the use of gene-editing technologies, including endorsing research into therapeutic applications of somatic cell gene editing; supporting the use of gene-editing standards to promote safe and effective gene-editing therapies; calling for the continued development of national and regional regulatory frameworks to guide the advancement of somatic cell gene editing; committing to a moratorium on the current use of germline gene editing in human clinical settings; and agreeing not to support or condone the use of germline gene editing in human clinical trials or human implantation unless ethical and potential safety concerns related to germline gene editing are adequately addressed. The statement was co-signed by 13 organizations utilizing gene-editing technologies, including prominent companies such as Bluebird Bio, which has marketed gene therapy products; Editas Medicine, founded by Chinese-American scientist Feng Zhang; and Sangamo Therapeutics, a dominant player in ZFN technology.
Payment System
Several gene therapies developed for genetic disorders have been successively approved for market launch, with their one-time treatment costs continuously shattering public expectations. The top three spots on the list of the most expensive drugs in history are all occupied by gene therapies: the most expensive is Novartis’s Zolgensma, at $2.125 million per dose; second is bluebird bio’s Zynteglo, at $1.8 million per dose; and third is Spark Therapeutics’ Luxturna, at $850,000 for both eyes.
Although the further approval of related drugs will break the market exclusivity of oligopolies and force them to lower prices, the high costs associated with the research and development, manufacturing processes, and post-treatment patient monitoring of gene therapy drugs themselves dictate that their prices will remain elevated in the short term. Therefore, how to pay for these high-cost drugs remains a significant challenge.
Current approaches include joint negotiations between payers and pharmaceutical companies, with the latter proposing innovative payment models such as installment payments, deferred payments, and outcome-based pricing.
For instance, Bluebird Bio has stated that the full price of Zynteglo can be paid in five annual installments of approximately $357,567. Patients are required to pay an upfront fee equivalent to 20% of the one-time gene therapy cost at the start of treatment, while the remaining 80% is contingent upon treatment success. Meanwhile, Novartis is also negotiating “outcome-based agreements” with insurers to establish payment models over a five-year period.
Gene therapy companies such as Novartis and Bluebird Bio have introduced value-based, targeted payment models, wherein full payment is contingent upon therapeutic efficacy or is disbursed in installments over several years. However, the current U.S. Medicaid and Medicare systems were not designed to accommodate these novel approaches, and patients continue to face multiple challenges regarding payment for gene therapies.
Therefore, payment reforms for gene therapies targeting rare diseases are urgently needed. We have reviewed the payment methods, policies, and restrictions associated with certain currently marketed gene therapy drugs for rare diseases, as detailed in the table below.
Overview of Payment Coverage for Gene Therapies Targeting Rare Diseases
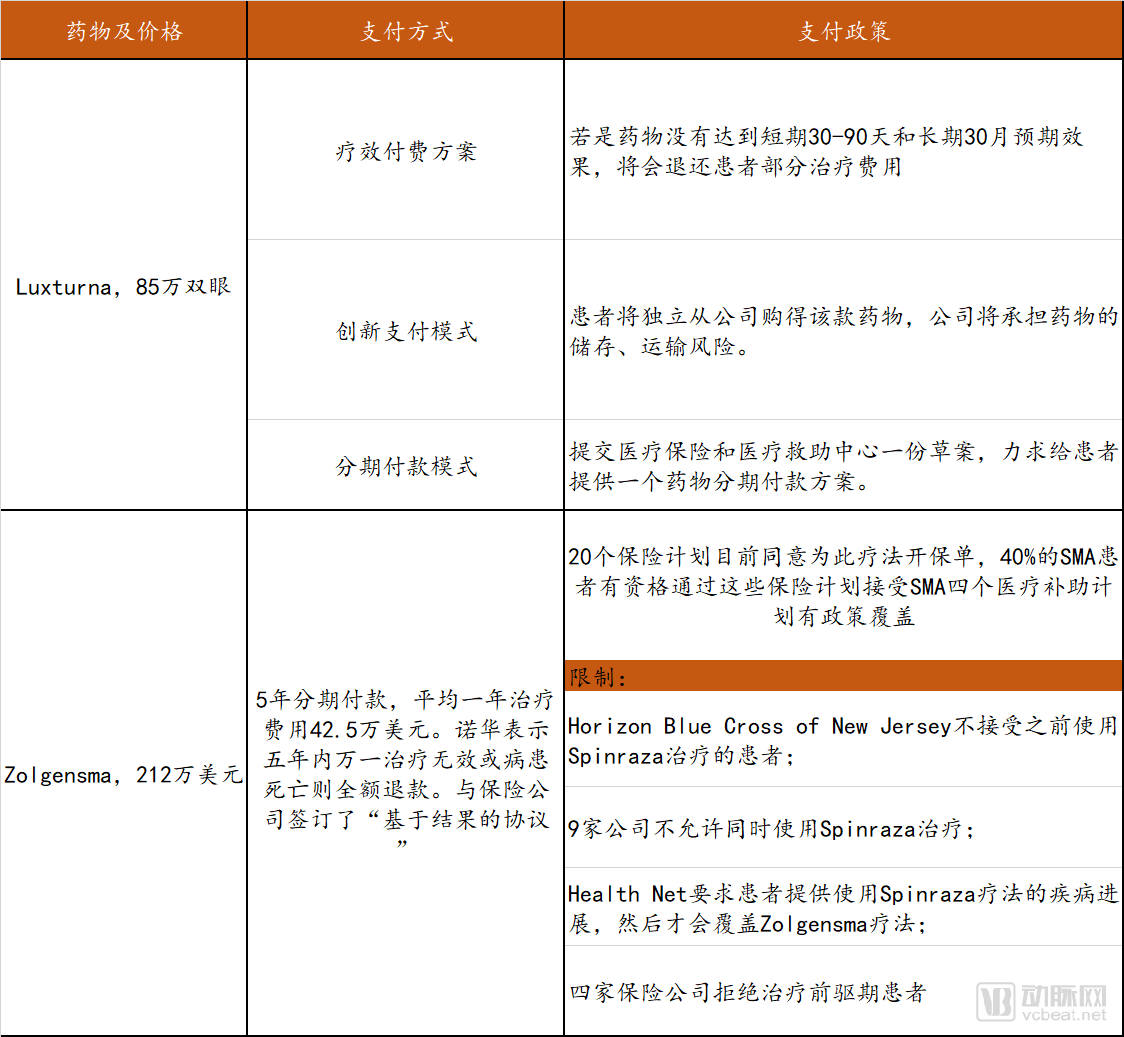
Data Source: FiercePharma, Probe Capital
Drawing on the rare disease coverage systems of various countries, the United States adopts a model in which commercial insurance serves as the primary payer, supplemented by government healthcare programs. Patients may voluntarily enroll in prescription drug plans through commercial insurers or pharmacy benefit managers, flexibly selecting options based on tiered formularies, varying copayment amounts, and reimbursement levels for drugs at different tiers. The healthcare security systems for rare diseases in other countries are illustrated in the figure below. Given the potential of rare disease gene therapies to achieve “one-time treatment, lifelong cure,” and assuming further reductions in therapy prices, gene therapy may ultimately impose a lower overall economic burden on society compared with conventional treatments. Therefore, the possibility of financial support from national governments or medical assistance programs in this area cannot be ruled out.
In summary, we speculate that the payment models for gene therapies targeting rare diseases may evolve from the current approaches—such as time-based fees, outcome-based pricing, and insurer-led payments—to a hybrid model featuring multiple payers and diverse payment mechanisms.
China’s rare disease coverage system is relatively singular compared to those of the aforementioned countries, relying primarily on reimbursement through basic medical insurance. There are few commercial insurance products in China that cover rare diseases, and those available often feature high premiums and stringent claims conditions. Several provinces and municipalities in China (such as Shanghai, Qingdao, and Zhejiang) are exploring and implementing locally tailored models for rare disease drug coverage. Overall, the future direction of China’s coverage system for gene therapies targeting rare diseases lies in strengthening medical insurance negotiations to include these therapies in the national reimbursement drug list, and establishing a multi-stakeholder, multi-source financing payment system.
Key References
1. Wang D, Tai PWL, Gao G. Nat Rev Drug Discov. 2019 May;18(5):358-378.
2. Anguela XM, High KA. Annu Rev Med. 2019 Jan 27;70:273-288.
3. High KA, Roncarolo MG. N Engl J Med. 2019 Aug 1;381(5):455-464.
4. Gaj T, Gersbach CA, Barbas CF 3rd. Trends Biotechnol. 2013 Jul;31(7):397-405.
5.Dunbar CE, High KA, Joung JK, Kohn DB, Ozawa K, Sadelain M. Science. 2018 Jan 12;359(6372).
6. Miller JC, Patil DP, Xia DF, Paine CB, Fauser F, Richards HW, Shivak DA, Bendaña YR, Hinkley SJ, Scarlott NA, Lam SC, Reik A, Zhou Y, Paschon DE, Li P, Wangzor T, Lee G, Zhang L, Rebar EJ. Nat Biotechnol. 2019 Aug;37(8):945-952.
7. Paschon DE, Lussier S, Wangzor T, Xia DF, Li PW, Hinkley SJ, Scarlott NA, Lam SC, Waite AJ, Truong LN, Gandhi N, Kadam BN, Patil DP, Shivak DA, Lee GK, Holmes MC, Zhang L, Miller JC, Rebar EJ. Nat Commun. 2019 Mar 8;10(1):1133
8. Miller JC, Tan S, Qiao G, Barlow KA, Wang J, Xia DF, Meng X, Paschon DE, Leung E, Hinkley SJ, Dulay GP, Hua KL, Ankoudinova I, Cost GJ, Urnov FD, Zhang HS, Holmes MC, Zhang L, Gregory PD, Rebar EJ. Nat Biotechnol. 2011 Feb;29(2):143-8
9. Wang H, La Russa M, Qi LS. Annu Rev Biochem. 2016 Jun 2;85:227-64.
10. Fellmann C, Gowen BG, Lin PC, Doudna JA, Corn JE. Nat Rev Drug Discov. 2017 Feb;16(2):89-100
11. Gammage PA, Viscomi C, Simard ML, Costa ASH, Gaude E, Powell CA, Van Haute L, McCann BJ, Rebelo-Guiomar P, Cerutti R, Zhang L, Rebar EJ, Zeviani M, Frezza C, Stewart JB, Minczuk M. Nat Med. 2018 Nov;24(11):1691-1695
12. Bacman SR, Kauppila JHK, Pereira CV, Nissanka N, Miranda M, Pinto M, Williams SL, Larsson NG, Stewart JB, Moraes CT. Nat Med. 2018 Nov;24(11):1696-1700