Ongoing Nitrosamine Contamination Crisis: Regulatory Responses from NMPA, FDA, and EMA Following the Valsartan and Ranitidine Incidents
On September 13, 2019, the U.S. Food and Drug Administration (FDA) issued a statement indicating that certain ranitidine medications (including well-known products for treating gastric ulcers such as Zantac) contained a nitrosamine impurity called N-nitrosodimethylamine (NDMA). On the same day, the Executive Director of the European Medicines Agency (EMA) also released a statement requesting guidance from the Committee for Medicinal Products for Human Use (CHMP) on methods to avoid nitrosamines in human medicines, and noted that nitrosamine impurities had been detected in samples of pioglitazone and ranitidine from one company. Currently, the FDA and EMA have initiated reviews of ranitidine within the United States and the European Union, respectively.
The NDMA impurity recently detected in ranitidine is no stranger; it was the central figure in the major controversy surrounding Huahai Pharmaceutical’s valsartan just one year ago.
In 2018, Huahai Pharmaceutical triggered investigations by the European Medicines Agency (EMA) and the U.S. Food and Drug Administration (FDA) after the impurity N-nitrosodimethylamine (NDMA) was detected in its valsartan active pharmaceutical ingredient (API). On July 5, the EMA issued a recall announcement, stating that NDMA had been detected in valsartan produced at Huahai’s Chuannan facility. Random spot checks revealed impurity levels ranging from 3.4 to 122 ppm, with an average of 66.5 ppm. According to the International Council for Harmonisation of Technical Requirements for Pharmaceuticals for Human Use (ICH) Guideline M7, which assesses and controls DNA-reactive (mutagenic) impurities in pharmaceuticals to limit potential carcinogenic risk, this level of impurity warranted concern. The FDA conducted an on-site inspection of Huahai’s Chuannan facility from late July to early August and, on September 28, placed the facility on Import Alert 66-40.
Based on the FDA warning letters, the author has compiled a timeline of the NDMA issue involving Huahai Pharmaceutical’s valsartan, as shown in the table below.
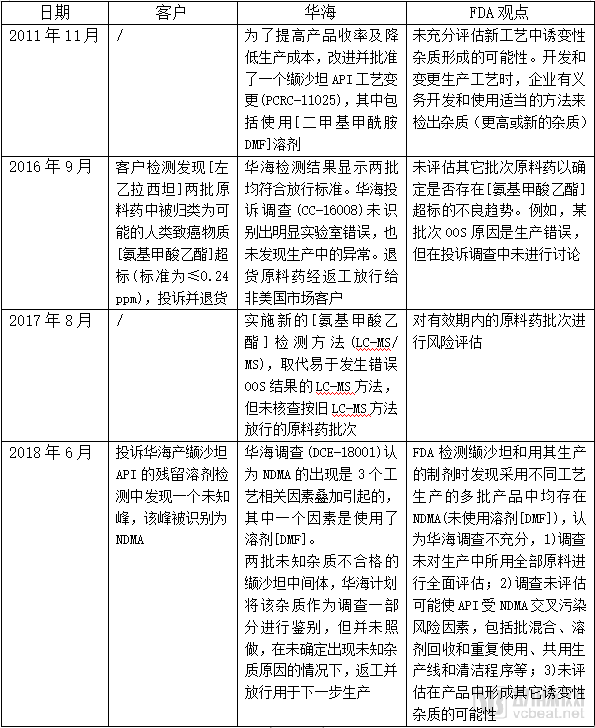
[ ]: Hidden information in the FDA warning letter; refer to Huahai Pharmaceutical’s translation documents for completion.
Huahai stated that when the NDMA impurity was discovered, regulatory authorities in various countries had not yet established industry standards for acceptable control limits for this impurity. Its current valsartan manufacturing process was approved by the EMA and FDA in 2012 and 2013, respectively, and complied with regulatory standards.
However, the FDA cautions that general industry practices may not always align with Current Good Manufacturing Practice (CGMP) requirements, and manufacturers remain responsible for the quality of their drug products. ICH M7 indicates that when changes are made to the active pharmaceutical ingredient (API) manufacturing process, the change assessment must determine whether such changes could lead to the formation of any new mutagenic impurities or result in higher levels of known mutagenic impurities. The FDA works closely with manufacturers to address impurity issues in products and to increase the supply of shortage drugs.
Nitrite Compounds
Prior to June 2018, NDMA and NDEA were not among the identified impurities in sartan drugs, and thus were not detected by routine testing. Some sartans contain a specific cyclic structure (a tetrazole ring), which may lead to the formation of nitrosamine impurities during synthesis under certain conditions when specific solvents, reagents, and raw materials are used. Furthermore, the use of contaminated equipment or reagents by manufacturers during the production process can also result in drug impurities. Other classes of drugs lacking the tetrazole ring, such as azilsartan, eprosartan, and telmisartan, are currently not associated with this risk.
The NMDA incident had a significant impact on Huahai Pharmaceutical. In 2017, Huahai Pharmaceutical’s sales revenue from valsartan active pharmaceutical ingredients (APIs) amounted to RMB 328 million, while sales of valsartan finished dosage forms reached USD 20.43 million. In 2018, the United States halted imports of valsartan APIs manufactured by Huahai, as well as finished products made from these APIs. The European Union also banned products from Huahai Pharmaceutical’s Chuan Nan factory in Linhai, Zhejiang Province, from entering its market. In addition to direct losses resulting from production halts and supply suspensions, Huahai faces product recalls, customer compensation claims, and potential subsequent litigation costs.
The sartan drug market, with a total market capitalization exceeding RMB 10 billion, was also shaken. Following Huahai, the detection of NDMA or NDEA in finished products from other companies such as Aurobindo, Mylan, Sandoz, and Teva triggered recalls of thousands of batches of valsartan, losartan, and ranitidine. This led to shortages of critical medications, including valsartan and losartan products, compelling the FDA to expedite and prioritize the review of new drug applications or generic drug applications aimed at alleviating these shortages. In March 2019, the FDA granted priority review and approval for the generic version of Diovan (valsartan) submitted by the Indian pharmaceutical company Alkem Laboratories Limited. The FDA evaluated the company’s manufacturing processes to ensure that appropriate testing methods were employed to demonstrate that the approved valsartan products did not contain NDMA or NDEA and posed no known risk of forming other nitrosamine impurities.
NDMA and NDEA (N-nitrosodiethylamine) are both nitrosamine impurities. They have been identified as animal carcinogens through animal studies and are currently classified as suspected human genotoxic carcinogens. The EMA cited a review article that assessed the maximum potential cancer risk associated with nitrosamine impurities (extrapolated from animal studies): among 100,000 patients taking the highest daily dose of Huahai Valsartan (calculated based on an average impurity content of 66.5 ppm), there would be 22 additional cases of cancer potentially attributable to NDMA after six years; for those taking the highest daily dose for four years, the number of cancer cases potentially attributable to NDEA would be eight.
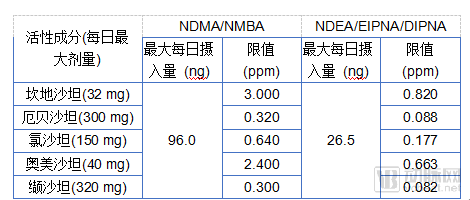
On January 31, 2019, the EMA recommended that manufacturers of sartan antihypertensive drugs review their manufacturing processes to avoid the formation of nitrosamine impurities and established interim temporary limits for NDMA and NDEA in accordance with current international guidelines. These limits were calculated by dividing the maximum daily intake of each impurity derived from animal studies (NDMA: 96.0 ng; NDEA: 26.5 ng) by the maximum daily dose of each active substance (see table below).
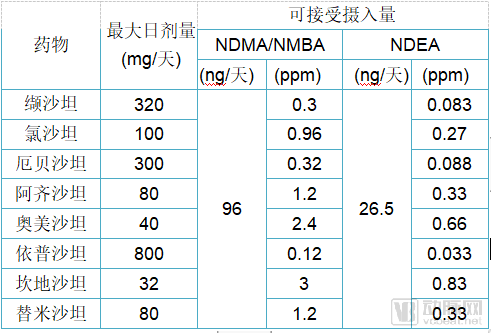
Subsequently, the FDA identified a new nitrosamine impurity, N-nitroso-N-methyl-4-aminobutyric acid (NMBA), in certain losartan products. Preliminary assessments indicate that the increased cancer risk for patients exposed to NMBA is similar to that associated with NDMA exposure but lower than the risk associated with NDEA exposure. On February 28, 2019, the FDA issued interim limits for NDMA, NDEA, and NMBA impurities in angiotensin II receptor blockers (ARBs) (see table below).
The testing methods for NDMA in sartan drugs, established by the Official Medicines Control Laboratories (OMCLs) of the European Union, are available on the website of the European Directorate for the Quality of Medicines & HealthCare (EDQM).NDEA Method*, and analytical methods for nitrosamine impurities in other APIs can be developed and validated based on these approaches.
On September 19 and 26, 2019, the European Medicines Agency (EMA) issued notices requiring marketing authorization holders (MAHs) for human medicines to review the risk of nitrosamine contamination in their products within six months. If nitrosamines are detected in a drug, MAHs must promptly notify the EMA to facilitate appropriate regulatory actions. High-risk products are prioritized, with the aim of resolving nitrosamine contamination issues within three years. The U.S. Food and Drug Administration (FDA) has also issued guidance on nitrosamine impurities in drugs such as sartans and ranitidine.Detection Method。
On January 2, 2019, the Chinese Pharmacopoeia Commission issued the “Public Notice on the Revised Draft of the National Standard for Valsartan (Second Edition),” proposing to add limits for N-nitrosodimethylamine (NDMA) and N-nitrosodiethylamine (NDEA) in the monograph for Valsartan (Volume II, Page 1547 of the 2015 edition of the Chinese Pharmacopoeia). The proposed limits are not more than 0.3 ppm and 0.08 ppm, respectively. On August 2, 2019, the Chinese Pharmacopoeia Commission again issued the “Public Notice on the Draft Revisions to the National Drug Standards for Valsartan, Irbesartan, and Telmisartan,” adding [Production Requirements] to require an assessment of the manufacturing process to determine the potential formation of genotoxic impurities such as NDMA and NDEA. The notice stated that, when necessary, appropriate analytical methods should be employed to analyze the product and confirm that the levels of NDMA, NDEA, and other related impurities comply with the relevant guidelines issued by China’s drug regulatory authorities or with the ICH M7 guideline.
The new version of the Drug Administration Law of the People's Republic of China, which came into effect on December 1, 2019, states in Article 28 that national drug standards refer to the Pharmacopoeia of the People's Republic of China and other drug standards promulgated by the drug regulatory department under the State Council. Where the drug quality standards approved by the drug regulatory department under the State Council are higher than the national drug standards, or where no national drug standards exist, the approved drug quality standards shall be applied. On November 20, 2018, the Chinese Pharmacopoeia Commission formulated the Administrative Measures for Research Projects on the Formulation and Revision of Drug Standards (Trial), encouraging enterprises to participate in the formulation and revision of drug standards. Henceforth, one of the sources of national drug standards will be the registration standards of drug manufacturers. As manufacturers possess extensive knowledge and understanding of their products’ foundational research and production processes, they can actively cooperate to enhance national drug standards and improve drug quality.
Following the valsartan incident, the EMA required MAHs and manufacturers to consider the following:
• Is there a risk of nitrosamine formation during the synthesis of the active pharmaceutical ingredient (API) due to the combination of reagents, solvents, catalysts, starting materials, formed intermediates, impurities, and degradation products?
• Is there a potential risk of nitrosamine contamination (from recycled materials, such as solvents, reagents and catalysts, equipment, degradation, starting materials or intermediates)?
• Is there a possibility of nitrosamine formation during the production of the finished product and/or storage throughout its shelf life?
The MAH and the manufacturer shall conduct validation by testing representative samples of relevant raw materials, intermediates, active pharmaceutical ingredients (APIs), or finished products. The number of batches/samples tested shall be scientifically reasonable and justified. Currently, the known pathways for nitrosamine impurities in drugs are as follows:
1. During the processing of active pharmaceutical ingredients (APIs), under certain processing conditions,Certain raw materials, starting materials, and intermediates are present, which may lead to the formation of nitrosamines, and these impurities are not completely removed in subsequent steps. The use of sodium nitrite (NaNO2) or other nitrites in the presence of secondary or tertiary amines is a potential cause of nitrosamine formation. In most confirmed cases of nitrosamine contamination in active pharmaceutical ingredients (APIs), nitrites and amines are used in the same step. However, another possibility exists: if sodium nitrite is used as a reagent in the preparation process of a certain step, despite extensive purification operations, residual amounts may be carried over into subsequent steps and react with amines to form nitrosamine impurities. Since the transfer of nitrites from one step to the next cannot be completely excluded, the risk of generating nitrosamine impurities should be considered for all processes involving the use of sodium nitrite (or other sources of nitrites) if amines are present at any stage of the synthesis.
2、Use of contaminated raw materials during the production processSubsequently, the recovery of solvents, reagents, and catalysts carries a risk of nitrosamine formation. Recovered materials (such as solvents, reagents, and catalysts) are typically outsourced to third-party vendors. In certain cases, these third-party recovery facilities do not receive sufficiently specific information regarding the materials they process and perform routine recovery procedures in non-dedicated equipment. If adequate cleaning of equipment between clients is not performed, or if preventive measures to avoid nitrosamine formation are not implemented, cross-contamination of solvents, reagents, and catalysts from different sources or processes may occur.
3、 Contaminated Starting Materials, including raw materials or intermediates, introduce nitrosamines.
4、 Degradation of Starting Materials, Intermediates, and Drug Substances, including degradation induced by reactions with residual NaNO2 or other nitrosating agents. This may also occur during the formulation or storage of the finished product.
5、Use specific packaging materials.I. The MAH observed nitrosamine contamination in the finished products within blister packs. It is hypothesized that the sealing aluminum foil, which contains nitrocellulose-based printing materials, can react with amines present in the ink to form nitrosamines, which then transfer into the product during specific packaging processes.
Author Biography

References
https://www.fda.gov/news-events/press-announcements/statement-alerting-patients-and-health-care-professionals-ndma-found-samples-ranitidine
https://www.fda.gov/inspections-compliance-enforcement-and-criminal-investigations/warning-letters/zhejiang-huahai-pharmaceutical-566685-11292018
https://www.ema.europa.eu/en/news/ema-provide-guidance-avoiding-nitrosamines-human-medicines
https://www.ema.europa.eu/en/medicines/human/referrals/angiotensin-ii-receptor-antagonists-sartans-containing-tetrazole-group
https://www.ema.europa.eu/en/documents/referral/nitrosamines-emea-h-a53-1490-information-nitrosamines-marketing-authorisation-holders_en.pdf
http://www.chp.org.cn/view/ff80808167f36c8401680c3d64812849?a=BZHXYP
http://www.chp.org.cn/view/ff8080816c2d76c5016c51c48ab54767?a=BZHXYP