Comprehensive Overview of FDA-Approved Innovative Drugs and First Generics in 2019: 38 NDAs, 10 BLAs, 96 First Generics, and 24 CBER Approvals
In 2019, the U.S. Food and Drug Administration’s Center for Drug Evaluation and Research (CDER) approved 48 New Drug Applications (NDAs) and Biologics License Applications (BLAs), as well as 96 first-generic drugs. Among the 48 initial NDA and BLA approvals, 37 were new molecular entities, 21 were orphan drugs, and 24 received priority review.
Under the Generating Antibiotic Incentives Now (GAIN) title of the FDA Safety and Innovation Act, the following new antibiotics have been granted Qualified Infectious Disease Product (QIDP) designation by the FDA: Fetroja (for the treatment of complicated urinary tract infections), Xenleta (for the treatment of community-acquired bacterial pneumonia), the novel anti-tuberculosis drug Pretomanid, Zerbaxa (for the treatment of hospital-acquired bacterial pneumonia and ventilator-associated bacterial pneumonia), and Recarbrio (a triple-combination injection of imipenem, cilastatin, and relebactam for the treatment of complicated urinary tract infections and complicated intra-abdominal infections in adults). The Global Alliance for TB Drug Development, which is developing Pretomanid, has also received a Priority Review Voucher for neglected tropical diseases.
Vyondys 53, the first targeted therapy for Duchenne muscular dystrophy, a rare disease caused by specific mutations, has been granted Priority Review and Orphan Drug Designation by the FDA. Additionally, the FDA awarded Sarepta Therapeutics Inc., the developer, a Rare Pediatric Disease Priority Review Voucher.
In light of the substantial benefits that the new cystic fibrosis drug Trikafta brings to a broad population of patients with cystic fibrosis, the FDA granted Trikafta all available designations—including Priority Review, Fast Track, Breakthrough Therapy, and Orphan Drug—to expedite its approval in the most efficient manner. The review was completed in just three months, nearly five months ahead of schedule. Meanwhile, the FDA awarded Vertex Pharmaceuticals a Rare Pediatric Disease Priority Review Voucher.
Genentech’s Rozlytrek (entrectinib, for the treatment of tumors harboring NTRK [neurotrophic receptor tyrosine kinase] gene fusions) is the third cancer therapy approved by the FDA based on a common biomarker across different tumor types rather than on the tissue of origin. This approval marks a new paradigm in “tissue-agnostic” cancer drug development. The other two drugs are pembrolizumab (approved in 2017 for the treatment of microsatellite instability-high [MSI-H] or mismatch repair-deficient [dMMR] tumors) and larotrectinib (approved in 2018 for the treatment of NTRK gene fusion-positive tumors).
Novartis’s Piqray (alpelisib) is the first PI3K inhibitor indicated for breast cancer and the first new molecular entity approved under the Real-Time Oncology Review (RTOR) pilot program. RTOR allows the FDA to begin analyzing key efficacy and safety datasets prior to formal application submission, enabling the review team to commence their assessment earlier and engage with the applicant sooner. Piqray also utilized the latest Assessment Aid (AAid), a multidisciplinary review template designed to focus the FDA’s written review on critical thinking and consistency while reducing time spent on administrative tasks. The approval of Piqray occurred approximately three months ahead of the Prescription Drug User Fee Act VI (PDUFA) target date.
The first oral GLP-1 drug, Novo Nordisk’s Rybelsus (oral semaglutide), has been approved. It is the first non-injectable glucagon-like peptide-1 (GLP-1) receptor agonist for the non-insulin treatment of patients with type 2 diabetes. Meanwhile, Eli Lilly’s Baqsimi nasal powder inhaler is the first approved glucagon therapy for the emergency treatment of severe hypoglycemia that does not require injection.
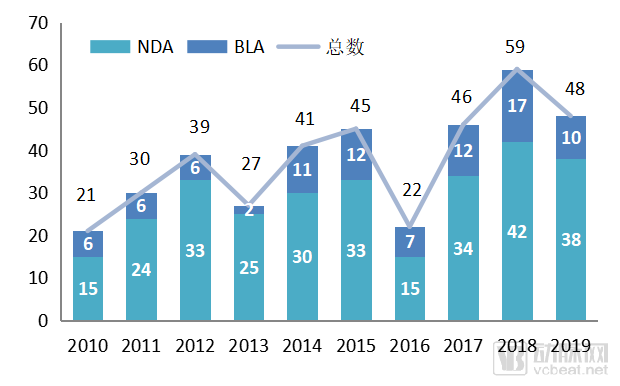
Figure 1. Number of New Drugs Approved by CDER, 2010–2019
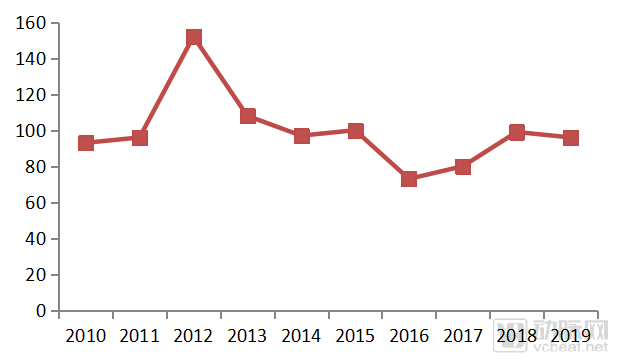
Figure 2. Number of First Generic Drugs Approved by CDER, 2010–2019
In February 2019, the FDA issued the “Competitive Generic Therapies (CGT) Guidance,” aiming to incentivize pharmaceutical companies to develop generic versions of drugs with insufficient competition through policy support. According to the FDA Reauthorization Act enacted in 2017, a drug is deemed to have inadequate generic competition if there is no more than one approved drug listed in the FDA’s “Orange Book.” Applications for generics of such drugs are eligible for CGT designation.
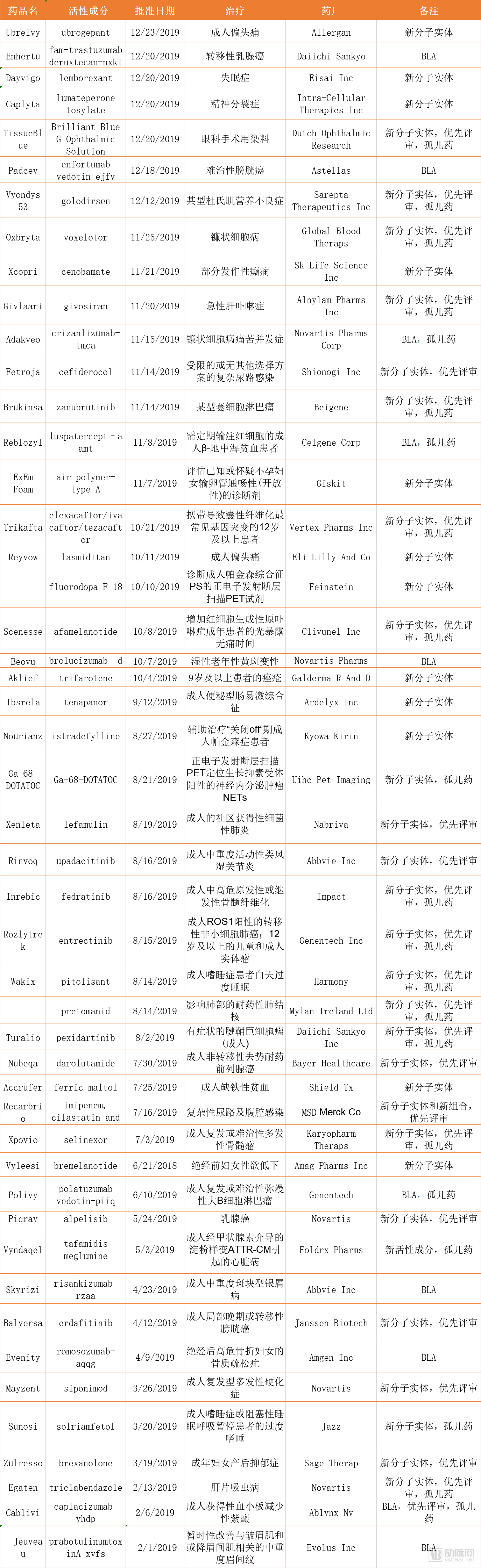
Table 1. 48 NDA and BLA Applications
In 2019, the FDA’s Center for Biologics Evaluation and Research (CBER) approved 21 Biologics License Applications (BLAs), as well as three New Drug Applications (NDAs) and Abbreviated New Drug Applications (ANDAs). These approvals included three vaccine products (the first live vaccine for the prevention of smallpox and monkeypox, the first vaccine for the prevention of Ebola virus disease, and a tetravalent live dengue vaccine), as well as gene therapies.Zolgensma。
The FDA granted priority review and breakthrough therapy designation to ERVEBO, the first vaccine for the prevention of Ebola virus disease, completed its evaluation of Ervebo’s safety and efficacy in less than six months, and awarded a Priority Review Voucher for tropical diseases to Merck, the developer of the vaccine.
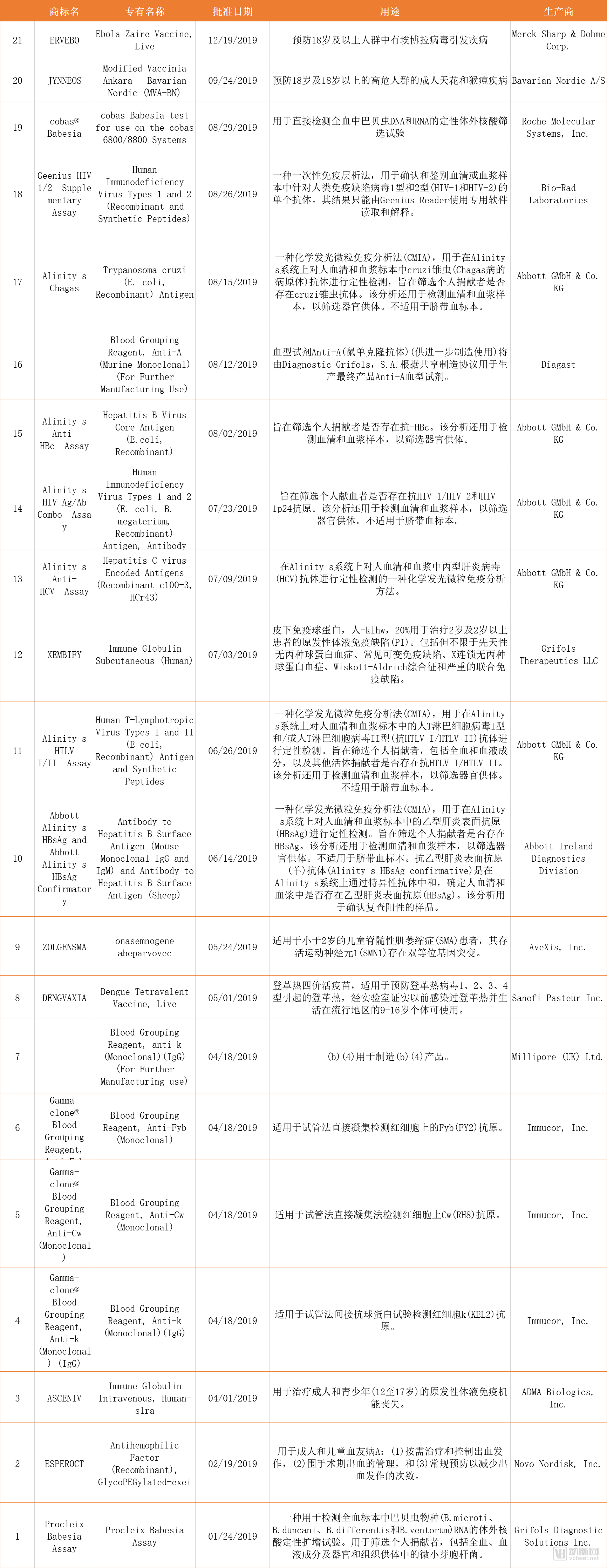
Table 2. Twenty BLA Applications
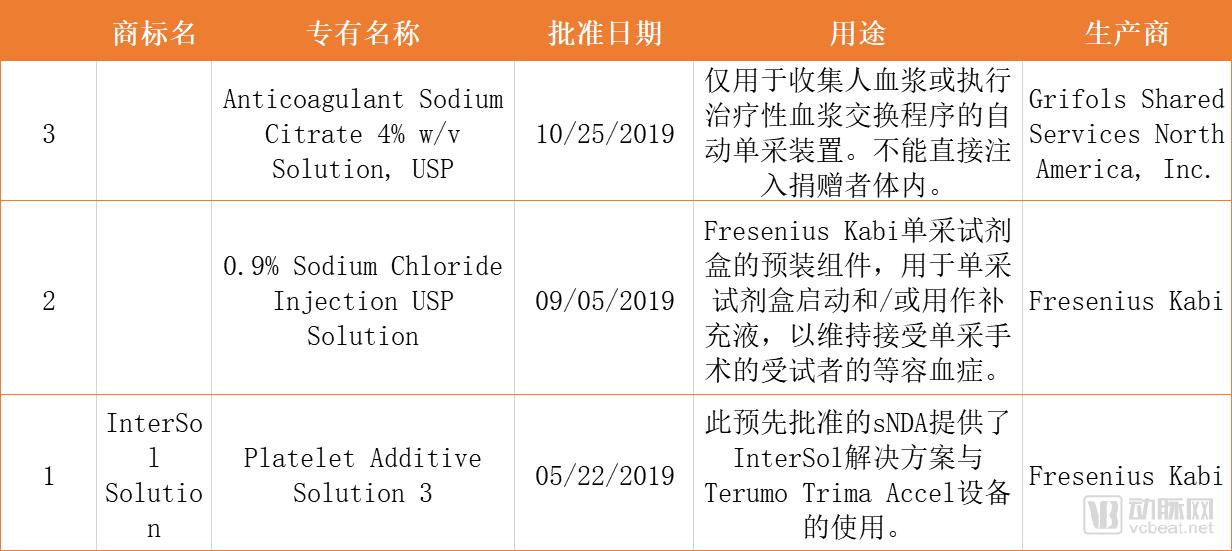
Table 3. Three NDA and ANDA Applications
About the Author
