NMPA Advances Real-World Evidence: Applications and Monetization of Clinical Big Data in Pharmaceutical Enterprises
On January 7, the National Medical Products Administration (NMPA) issued Document No. 1 of 2020, titled “Guiding Principles for Using Real-World Evidence to Support Drug Development and Regulatory Review (Trial),” which aims to clarify the definitions of real-world evidence in drug development and regulatory decision-making, provide guidance on the collection and applicability assessment of real-world data, and offer reference recommendations for the pharmaceutical industry and regulatory authorities in leveraging real-world evidence to support drug development and regulatory decisions.
Real-world studies refer to the research process of collecting health-related data on study subjects (real-world data) or aggregated data derived from these data in a real-world setting, aiming at pre-specified clinical questions, and obtaining clinical evidence on drug utilization and potential benefit-risk profiles (real-world evidence) through analysis. Currently, clinical big data and real-world studies are highly popular; however, in practice, there are challenges such as unclear business models and difficulties in achieving profitability.
As a series of reform measures are implemented, drug research and development (R&D) is advancing rapidly, with new drugs accelerating their market entry. This trend has also raised higher demands for the quality and efficiency of drug R&D efforts. Next, we will explore how pharmaceutical companies are applying and monetizing clinical big data, as well as the application scenarios of real-world studies.
Unlike pharmaceutical or health data, clinical big data involves extremely high barriers to acquisition, which inherently endows the data with significant value. In other words, whoever possesses the data holds the absolute initiative in developing clinical data applications. Currently, clinical big data primarily serves four sectors: government, pharmaceutical companies, hospitals, and insurance providers. While these four user categories are relatively representative, this article focuses exclusively on discussing the application and monetization models of clinical big data within pharmaceutical companies. It is worth noting that these sectors frequently interact; for instance, the pharmaceutical company-hospital relationship represents a typical collaborative system. A complete business model is formed only when clinical big data serves both pharmaceutical companies and hospitals simultaneously.
Pharmaceutical companies are key beneficiaries of clinical big data, with application scenarios primarily falling into two categories: facilitating drug R&D and enhancing pharmaceutical marketing. Given their relatively strong financial resources, pharmaceutical companies are keen to adopt clinical big data products or services that deliver value in these two areas.
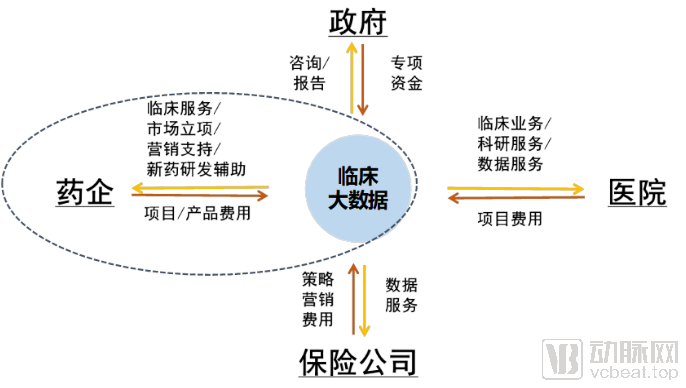
Clinical big data services also revolve around two aspects: drug R&D and drug marketing.
There is a wide range of activities that can be undertaken during the drug development process. Drug development is generally divided into three phases: preclinical, clinical, and post-marketing. As this article focuses exclusively on the application of clinical big data, the preclinical phase falls outside the scope of discussion. In the clinical phase, since a substantial portion of research is prospective in nature, data must be collected and analyzed based on trial design protocols. While existing clinical data have limited utility for conducting prospective studies, they are highly effective for identifying and recruiting eligible patients. Furthermore, clinical data serve as robust evidence to support drug project initiation decisions, including whether to launch new drug development programs or to pursue in-licensing opportunities for new varieties.
During the drug marketing phase, clinical data finds extensive application in areas such as post-marketing evaluation, real-world studies (RWS), marketing knowledge graphs, and health monitoring and follow-up. For instance, robust evidence for a specific drug derived from real-world studies can not only be published publicly but also incorporated into the drug’s package insert. These applications offer substantial benefits to pharmaceutical companies’ marketing efforts.
I. Drug Development
Drug development is divided into two phases: preclinical research and clinical research, as shown in the figure below. Preclinical research is primarily conducted at the laboratory stage, where existing clinical data have limited applicability. The clinical phase of new drug development mainly involves prospective studies, i.e., recruiting a cohort of patients to participate in trials aimed at achieving specific clinical endpoints. In drug clinical trials, nearly all data are newly generated; therefore, our existing clinical data hold little direct value for the conduct of these new drug trials themselves. However, given the stringent eligibility criteria for trial enrollment, existing clinical data can assist research institutions in identifying suitable candidates for participant recruitment.

Clinical Trial Enrollment
Clinical trial enrollment involves multiple approaches, with the following being the primary methods currently in use:
(1) Ensure that subjects meet all inclusion criteria specified in the latest ethically approved protocol and do not meet any of the exclusion criteria;
(2) Recruitment advertisements: Recruitment advertisements that have received written approval from the Ethics Committee may be posted;
(3) Universal screening for all patients seeking medical care;
(4) Wait in the outpatient clinic for eligible subjects to present for consultation;
(5) Establish specialized clinics for specific medical specialties and diseases.
These approaches are essentially recruitment efforts that lack precision and suffer from low efficiency. Leveraging clinical data enables more accurate retrieval of patient-related information, thereby improving the efficiency of subject recruitment. However, due to the poor interoperability of electronic medical record (EMR) data in China, it is difficult to integrate EMRs within regions or standardize terminology, resulting in fragmented data that is challenging to search.
II. Pharmaceutical Marketing
1. Real-World Study
Post-Marketing Drug MarketingFollowing a drug’s market launch, the most critical issue becomes pharmaceutical marketing. The primary benefit of post-marketing clinical studies and real-world evidence (RWE) research to pharmaceutical companies lies in enhancing the market position of their products. Pharmaceutical companies invariably design their research agendas to yield favorable outcomes; should unfavorable evidence emerge, they proactively adjust corporate strategies to minimize potential losses. Therefore, from the perspective of corporate interests, these topics are discussed within the context of pharmaceutical marketing.
However, real-world studies are not limited to pharmaceuticals; there are also corresponding research pathways for controlling healthcare costs and improving healthcare quality. So why are there currently so many real-world studies related to drugs?
The reason is simple: research activities supported by a viable business model constitute a sustainable framework. Since drug-related research is directly tied to the commercial interests of pharmaceutical companies, these firms bear the associated costs. Consequently, the majority of real-world study projects currently available on the market are related to pharmaceuticals.
Another major direction of real-world studies is healthcare cost containment. Given the significant financial pressure on the national medical insurance system, the government has allocated substantial research funding to control these costs. However, in the realm of medical insurance cost containment, real-world studies have not yet established a clear business model with commercial insurance companies; consequently, the scope of research applications is less extensive than that for pharmaceuticals. Topics related to clinical big data and insurance will be discussed in future articles.
There are two different classification approaches to real-world studies in academia.
The first viewpoint holds that all retrospective studies fall under the category of real-world studies. In short, any research conducted on data generated from real-world clinical practice or adjacent healthcare industries is considered a real-world study. Such data are naturally produced during the course of diagnosis and treatment, rather than being obtained through artificially designed experiments.
The second perspective holds that real-world studies encompass all research outside the scope of traditional clinical medical research. Traditional medical research refers to studies beyond the conventional clinical study types found in textbooks, such as randomized controlled trials, cohort studies, case-control studies, and cross-sectional studies.

In light of the two aforementioned academic perspectives, the author is more inclined toward the first. In the author’s view, the essence of real-world study lies in the source of data. Data obtained through rigorous screening of study subjects (such as in traditional clinical trials) cannot be classified as real-world study. Only data collected on a non-intentional basis can be termed real-world study.
Therefore, real-world studies do not have selection criteria for study subjects; all data are generated entirely in real-world settings. In the FDA’s guidance, the definition of “real-world data” emphasizes two key points: routinely collected and from a variety of sources.
(1) Superiority, Equivalence, and Non-inferiority Studies
Verification of drug efficacy is one of the projects that pharmaceutical companies are most interested in. In order to verify the position of their drugs in the medical environment, pharmaceutical companies are willing to invest a lot of resources to carry out real-world research work. The so-called superiority trial is to verify whether a drug is better than another; equivalence trial, which tests whether one drug has the same effect as another; non-inferiority trial, which verifies that one drug is not inferior to another.
Studies on drug superiority, equivalence, and non-inferiority fall within the scope of traditional research and can be conducted through experimental design. However, in essence, such studies are more suited to real-world research. Only data generated from actual clinical practice can provide a robust objective evaluation.
Generally, for such trials, a widely recognized drug on the market should be selected as the positive control. By comparing with this drug, results related to superiority, equivalence, or non-inferiority can be obtained. Since the selected positive control drugs are usually strong competitors, all companies naturally hope to achieve superior results.
For superiority trials, the hypothesis test is:
Null Hypothesis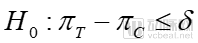
Alternative Hypothesis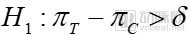
where δ is a clinically meaningful value, referred to as the superiority margin. πTTo evaluate the overall efficacy rate of the drug, πCOverall response rate of the control drug. The primary objective of a superiority trial is to demonstrate that the therapeutic efficacy of the investigational drug exceeds the prespecified superiority margin relative to the control drug, and that this difference is not less than zero.
For equivalence trials, the hypothesis testing is:
Null Hypothesis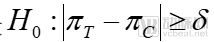
Alternative Hypothesis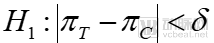
The relevant metrics in the formula are consistent with those mentioned earlier. In equivalence trials, if the difference in efficacy between the test drug and the reference drug falls within a predefined margin, statistical equivalence between the two can be established. Equivalence testing is more commonly applied in the consistency evaluation of generic drugs versus their originator counterparts.
For non-inferiority trials, the hypothesis test is:
Null Hypothesis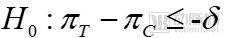
Alternative Hypothesis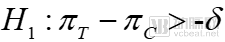
Non-inferiority trials focus on determining that the difference in efficacy between the investigational drug and the control drug is not less than the lower margin; a one-sided test can establish non-inferiority. The figure below vividly illustrates the relationship among three types of trials. If the efficacy of the investigational drug falls below the lower bound of the control drug’s efficacy, no conclusion can be drawn.
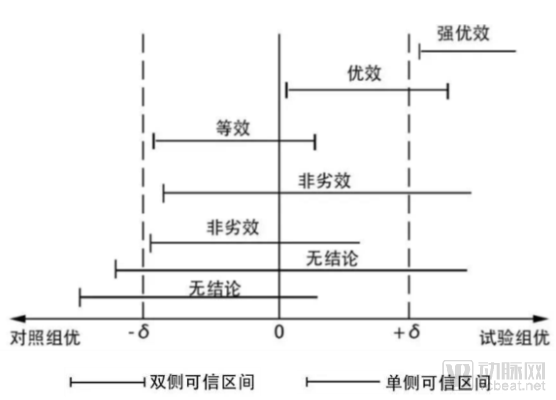
In traditional biostatistics, the aforementioned three types of trials can be conducted through methods such as deliberate study design and participant recruitment. For real-world studies, however, it is essential to rely exclusively on data generated in real-world settings, rather than utilizing data from artificially designed trials.
There are two main challenges in completing the above tasks: first, how to select the data, and second, how to define the clinical metric δ.
First, as discussed in the second question, defining the clinical margin δ has always been a challenge. If δ is set too large, it will widen the confidence interval, potentially reducing the demonstrated superiority of the investigational drug or leading to inequivalent drugs being incorrectly judged as equivalent. Conversely, if δ is set too small, it increases the likelihood of the opposite error. The specific determination of δ should be based on factors related to the drug’s indication. Commonly used metrics include biochemical markers and bioequivalence (BE), among others.
The first question is how to select data, which is crucial for real-world studies. There are two scenarios for data selection: one involves conducting retrospective research using existing medical data, while the other involves obtaining data through follow-up of populations taking two different drugs, constituting a prospective study.
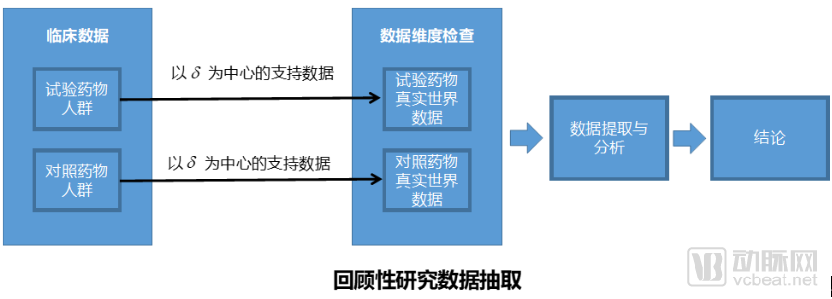
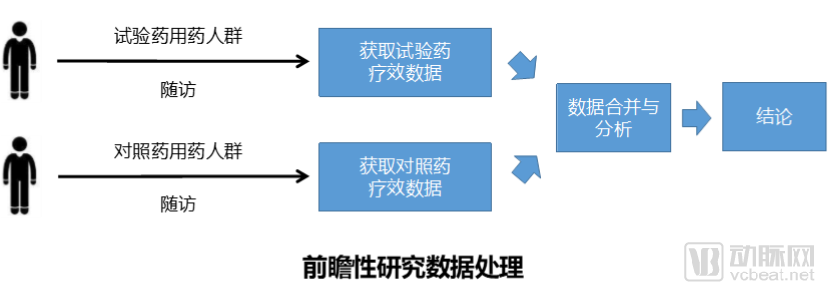
From the perspective of pharmaceutical companies, there is a strong desire to reach conclusions rapidly, which not only helps control costs but also benefits drug marketing and promotion. The challenge with retrospective studies lies in the fact that they rely on pre-existing clinical data, which is often of poor quality and difficult to obtain, potentially failing to meet the intended study objectives. Conversely, prospective studies require long follow-up periods and incur substantial costs, making it difficult for pharmaceutical companies to sustain such projects that demand significant investment without delivering timely results.
The greatest challenge currently facing clinical data is data transformation (ETL) and integration, which can also be considered part of data governance. In retrospective real-world studies, regarding what constitutes valuable clinical data, we offer three recommendations:
1. Clarify the sequence of clinical events;
2. Clarify patient identification;
3. Adopt unified coding standards.
Clarifying the sequence of clinical events is essential for determining a patient’s disease course and management. The underutilization of large volumes of clinical data stems from the inability to track a patient’s clinical interventions and corresponding outcomes over time. The application of clinical data does not necessarily require patients to be observed within the same time period, but it does necessitate clear definition of sequential factors and time intervals.
Clearly identifying patient identifiers is straightforward. Most real-world studies are conducted on a per-patient basis; even when investigating a specific drug or therapy, analysis must be performed at the individual patient level.
Adopting a unified clinical coding standard is a long-standing issue in the application of healthcare big data. Extensive discussion will not be provided here; it is hoped that all hospitals will strive to comply with this standard. The three basic requirements mentioned above are illustrated in the figure.
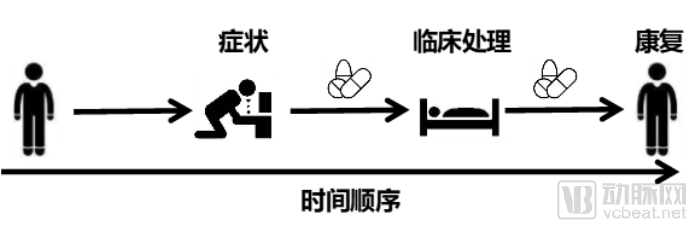
For real-world study findings, pharmaceutical companies should incorporate favorable conclusions into their product labeling, while promptly adjusting their market strategies to mitigate the impact of unfavorable results.
(2) Adverse Drug Reaction Monitoring and Pharmacovigilance
Adverse Drug Reaction Monitoring: More of a Responsibility and Obligation. In many foreign countries, much of the adverse reaction monitoring is carried out by pharmaceutical companies, whereas in China, it is primarily conducted by the government and relevant authorities.
Adverse drug reaction monitoring is primarily a corporate responsibility, particularly after a new drug is launched. It enables the refinement of package inserts based on monitored data, thereby enhancing medication safety. From the perspective of pharmaceutical companies’ interests, they should proactively conduct adverse drug reaction monitoring to mitigate the substantial compensation costs arising from such reactions.
The mechanisms underlying adverse drug reactions are relatively complex and can be broadly categorized into two major types: Type A and Type B.
The former is caused by an enhancement of the pharmacological effects of the drug. It is characterized by predictability and is generally dose-dependent. Although its incidence in the population is high, the mortality rate is low. The latter is an abnormal reaction completely unrelated to normal pharmacological effects, which is usually difficult to predict and cannot be detected through routine toxicology screening.
Common epidemiological evaluation methods include retrospective and prospective studies, which correspond to case-control studies and cohort studies, respectively. The former investigates the likelihood that a specific drug caused an adverse reaction after the reaction has already occurred; the latter involves following study subjects over a period of time to compare whether the incidence of adverse reactions is higher among those exposed to the drug than among those not exposed. Adverse drug reactions are often identified from real-world data and confirmed through traditional clinical trials and other methods.
Adverse drug reactions (ADRs) and pharmacovigilance have been extensively studied worldwide for many years. Various algorithms have been proposed, and data analysis approaches have evolved from focusing solely on healthcare data to integrating both healthcare and social media data. Currently, big data, machine learning, and other advanced algorithms are widely applied in the detection of ADR signals. In the field of ADRs and pharmacovigilance, two core tasks stand out: the construction of ADR databases and the mining of adverse reaction data.
The Adverse Drug Reaction (ADR) Database is a multi-source database, meaning it does not rely solely on data generated in clinical settings but also incorporates drug-related data such as protein and target information. Currently, most ADR data are derived from spontaneous reporting systems. In future developments, artificial intelligence and other technological approaches can be leveraged to construct a database of suspected adverse drug reactions. This database would draw directly from data generated during clinical processes, actively identifying ADR events by correlating clinical symptoms with medication usage patterns.
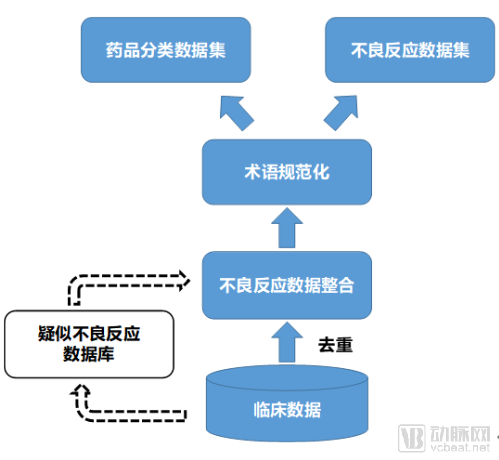
Research on algorithms for adverse drug reactions is also highly advanced. It is primarily categorized into three major types: Disproportionality Analysis (DPA), Logistic Regression Models (LRM), and Association Rule Mining (ARM).
In the current era of AI and machine learning development, algorithms such as decision trees, clustering, and neural networks are employed for the mining of adverse drug reactions. The specific details of these algorithms will not be elaborated upon in this article.
There are four experimental methods for exploring adverse drug reactions: the method of difference, the method of agreement, the method of concomitant variation, and the method of analogy. These four methods can also serve as the theoretical basis for clinical data research.
The Method of Difference involves identifying differing factors within similar contexts; such differing factors may potentially be the cause of a medical event. For example, in a patient population with arrhythmia, when traditional antiarrhythmic drugs proved ineffective, the medication was discontinued and amiodarone was subsequently administered. As a result, while the original arrhythmia remained uncontrolled in some patients, they additionally developed torsades de pointes. Within this same cohort, the identifiable differing factor before and after the use of amiodarone is precisely amiodarone itself. Therefore, it can be hypothesized that amiodarone may induce torsades de pointes. Based on this hypothesis, further analytical studies can be conducted.
The case-crossover method is applicable when the same medical event occurs in certain individuals across different times, locations, or demographic groups; this approach can be used to generate hypotheses. For example, this method can be employed in investigations of food poisoning.
The method of concomitant variation applies when the frequency of a medical event changes proportionally with variations in the quantity of an objective factor; such a factor may thus be considered a potential cause of the medical event. The globally sensational “Thalidomide Incident” exemplifies hypothesis generation through this approach, where researchers ingeniously correlated thalidomide’s market sales data from relevant years with medical events, plotting a time-distribution curve of total sales against the number of cases.
The analogy method involves comparing a medical event of unknown etiology with another well-understood objective factor; if similarities exist, it suggests that this objective factor may be the cause of the medical event. For example, in research on Reye’s syndrome, researchers observed that the clinical and histological changes associated with salicylate poisoning closely resembled those of the syndrome, leading to the formulation of a hypothesis through logical reasoning.
Pharmaceutical companies can leverage clinical data to conduct proactive research on adverse drug reactions (ADRs). The first step is to identify the relevant patient populations using the company’s specific drugs. Clinical phenomena observed within these populations are then clustered and categorized based on diagnostic and treatment event data. The findings from ADR research are typically presented in the form of a platform or system. Such platforms enable searches for adverse events, drugs associated with adverse events, and combinations of related adverse events.
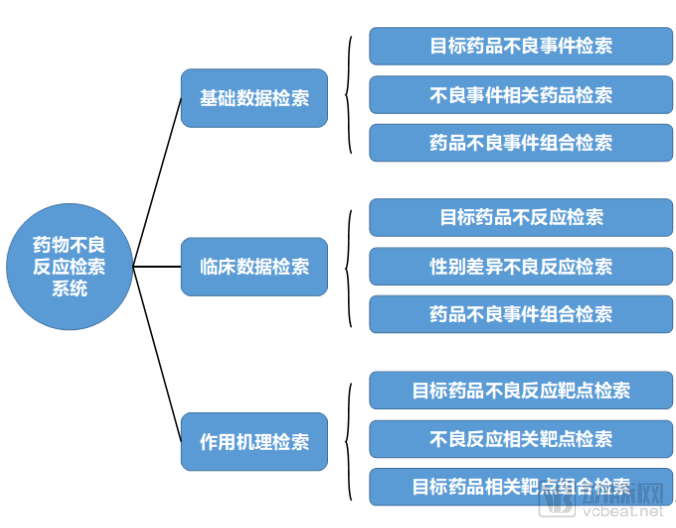
2. Drug Information Knowledge Graph
Previously, pharmaceutical marketing relied on medical representatives. However, with hospitals now imposing strict regulations on medical representatives, major pharmaceutical companies have increasingly shifted toward expert-driven marketing strategies. This approach promotes their products by educating physicians and patients, thereby conveying marketing messages through knowledge dissemination. Today’s medical representatives often carry tablets containing comprehensive drug information, relevant clinical evidence, and even pharmaceutical question-and-answer systems. Such a platform for retrieving pharmaceutical knowledge or a Q&A system can be built using knowledge graph technology.
For pharmaceutical companies, medical knowledge graphs represent one of the most direct applications in marketing. They also serve as a major application scenario for academic searches related to clinical and preclinical drug development.
The construction of medical knowledge graphs can be summarized into five components: representation, extraction, fusion, reasoning, and quality assessment of medical knowledge. By extracting constituent elements of knowledge graphs—such as entities, relationships, and attributes—from large volumes of structured or unstructured medical data, these elements are stored in a knowledge base using reasonable and efficient methods. Medical knowledge fusion disambiguates and links the content within the medical knowledge base, enhancing its internal logical consistency and expressive power. Furthermore, it updates outdated knowledge or supplements new knowledge to the medical knowledge graph through manual or automated approaches.
The data sources for knowledge graphs are highly diverse; clinical big data represents only one aspect of these sources. The figure below illustrates the diversity of data sources for knowledge graphs.

From a monetization perspective, knowledge graphs primarily deliver services in the form of knowledge bases, which can naturally be extended to business applications such as Q&A chatbots. Within enterprises, knowledge graphs can enhance pharmaceutical companies’ marketing capabilities, assisting sales representatives in promoting products more effectively. Externally, a knowledge graph functions as a comprehensive knowledge base system, enabling monetization through annual subscription fees charged for user accounts.
This article primarily focuses on the application and monetization of clinical big data in pharmaceutical companies. Unlike hospitals, the ultimate goal of enterprises is profitability; therefore, pharmaceutical companies will inevitably integrate clinical big data into their business models. The adoption of clinical big data by pharmaceutical firms serves as a positive driving force. Only business models that generate commercial benefits are sustainable, and such models will elevate the application of clinical data to new heights.
Author: Gao Fei (pen name: Bai Bai)
Senior Product Expert. Possesses an interdisciplinary background in mathematics, computer science, and pharmacy. Specializes in integrating industry insights with technology within the fields of big data and artificial intelligence, with over seven years of experience in AI algorithms and product management. Has conducted in-depth research on business operations and models related to the Industrial Internet.
Formerly served as a Senior Scientist at the Institute of Chemistry, Chinese Academy of Sciences, and Pharmaron, engaging in small-molecule drug research and development. Since 2014, has been dedicated to AI algorithm research and product development, leading the creation of China’s first intelligent preclinical drug data platform, which received high recognition from the Center for Drug Evaluation (CDE). Has repeatedly led the development of regional electronic medical record and health archive big data platforms, serving provinces such as Jiangxi and Shandong. Was invited to chair the Medical and Brain Science Division at the Science Magazine China Annual Conference. Was also invited to participate in the “PUMCH Centennial” information planning project at Peking Union Medical College Hospital, providing design solutions for an intelligent clinical research platform.
Current Director of the National Information Technology Committee for Chronic Disease Prevention and Control, Senior Member of the Chinese Pharmaceutical Association, and Executive Director of the China Health Information Association.