InferVision Secures FDA Approval for First Deep Learning-Based Pulmonary AI, Marking a Milestone for Chinese AI Companies Going Global

Infervision
Artificial Intelligence Product Developer
VCBeat learned that on July 3, 2020, Infervision’s AI-powered lung imaging product successfully received FDA clearance. This marks the first FDA-approved lung auxiliary detection software in the United States to utilize deep learning algorithms, representing a substantial breakthrough for Chinese medical AI companies expanding into overseas markets.

To gain deeper insights into the FDA approval process, VCBeat contacted Infervision’s regulatory affairs team. By examining this company’s application procedures, we may derive valuable lessons to help accelerate the approval of more AI-based medical products.
Since 2018, we have frequently discussed the overseas markets of Chinese AI medical imaging companies. In fact, there are quite a few AI enterprises that cultivate the domestic market while expanding abroad. However, regardless of their location, they all face the same challenge of medical device certification. The difficulty lies in the fact that many AI companies lack relevant approval experience.
In the United States, the FDA shares similarities with China’s NMPA; however, the two agencies differ significantly in their regulatory frameworks, approval pathways, and review timelines.
Specifically, both China's NMPA and the U.S. FDA conduct review and regulation of medical devices based on risk. The NMPA classifies medical devices into Class I, Class II, and Class III, while the FDA categorizes them into Class I, Class II, and Class III.
For Class I medical devices, after the enterprise submits the relevant documentation to the FDA, the FDA only issues a public notice and does not issue any certification to the enterprise. For Class II and Class III medical devices, enterprises must submit a 510(k) premarket notification or a Premarket Approval (PMA) application. Upon public announcement, the FDA will issue a formal market access clearance letter to the enterprise, after which the enterprise may sell its products in the U.S. market. Whether the FDA conducts an on-site assessment of the enterprise’s quality management system during the application process is determined comprehensively by the FDA based on factors such as product risk classification, regulatory requirements, and market feedback.
The NMPA categorizes medical devices into three distinct classes with varying regulatory requirements. For Class I devices, domestic manufacturers are required to complete registration filing and production licensing filing with the municipal-level drug administration in their locality. For Class II devices, domestic manufacturers must submit registration applications, undergo product testing (conducted by nationally accredited testing institutions), perform clinical evaluations (implementation depends on regulatory requirements), and pass quality management system audits with the provincial (or municipality directly under the Central Government) drug administration in their locality. For Class III devices, domestic medical device manufacturers must submit registration applications, undergo product testing (conducted by nationally accredited testing institutions), conduct clinical trials/evaluations (clinical trial waivers may be applied for if stipulated), and pass quality management system audits with the National Medical Products Administration (NMPA).
Generally, the FDA classifies imaging-based products as Class II and invasive products as Class III. Class II clearance is further categorized into De Novo, 510(k), and PMA pathways, depending on whether predicate devices exist and whether the specific risks are sufficiently understood and controllable. In China, AI products used for clinical decision support must obtain Class III certification.
Statistics from January 2018 to December 2019 show that more than 35 AI products received regulatory clearance, primarily through pathways such as De Novo and 510(k). However, these cleared products were predominantly non-diagnostic aid devices. Only a few, such as IDx’s IDx-DR and Aidoc’s CT-based solutions, closely resembled what is defined in China as “diagnostic aid” products. Moreover, none of the approved products were associated with Chinese companies.
Therefore, Infervision’s AI lung product receiving FDA approval can be regarded as a milestone for Chinese AI companies expanding overseas. Following the FDA 510(k) clearance of ClearRead CT, Infervision will be able to accelerate its commercialization efforts, and with the experience gained from this initial approval, additional products are expected to follow in quick succession.
To discuss the significance of regulatory approval, one must first address the intrinsic value of AI products. AI products cannot survive in isolation from the market, and Infervision has strategically targeted the substantial demand for lung cancer screening in the United States.
According to data provided by Infervision, lung cancer is the second most common cancer in the United States and one of the leading causes of cancer-related mortality, accounting for approximately 25% of all cancer deaths. The U.S. National Institutes of Health (NIH) projected that in 2020, there would be 135,720 lung cancer deaths nationwide, representing 22.4% of all cancer deaths, with a five-year survival rate of 20.5%.
Against the backdrop of persistently high and annually rising lung cancer incidence, the United States launched the Lung Cancer Screening (LCS) program. This initiative targets asymptomatic individuals at high risk for lung cancer, employing low-dose computed tomography (LDCT) for pulmonary imaging to facilitate early detection, diagnosis, and treatment, thereby improving survival rates.
Currently, the U.S. Centers for Medicare & Medicaid Services (CMS) has incorporated Lung Cancer Screening (LCS) into its insurance coverage system. This indicates that U.S. payers are incentivizing healthcare institutions to acquire AI systems to improve lung cancer detection rates, enabling early intervention at the initial stages of the disease. This approach will significantly enhance patient survival rates and substantially reduce healthcare expenditures.
Therefore, for Infervision, obtaining approval means that they can not only engage in commercial collaborations with more than 1,700 ACR-registered hospitals, imaging centers, and upstream and downstream enterprises in the United States that provide LCS services to explore business opportunities in LCS projects, but also offer AI support to various medical institutions and imaging centers in North America for lung disease screening programs.
In fact, Infervision has established extensive collaborations with numerous top-tier medical institutions in North America, including prestigious U.S. healthcare providers such as the University of Maryland Medical Center and Jefferson Hospital. Following certification, Infervision’s business expansion efforts are poised to accelerate further.
Recalling the entire approval process, an Infervision representative could hardly contain their excitement: “Nine months is hardly a long time. We had expected to receive the certificate in another month or so, but unexpectedly, the FDA suddenly brought us good news.”
After completing U.S. clinical trials in October 2019, Infervision formally submitted its FDA 510(k) clearance application. “The process was not without challenges. Two months after submission, the FDA raised several issues regarding our documentation. In February, amidst the raging pandemic, part of our team was dedicated to developing AI products for COVID-19, while another portion worked on preparing supplemental materials for the FDA review. Both tracks progressed favorably, and we ultimately obtained clearance for our AI pulmonary nodule product in July.”
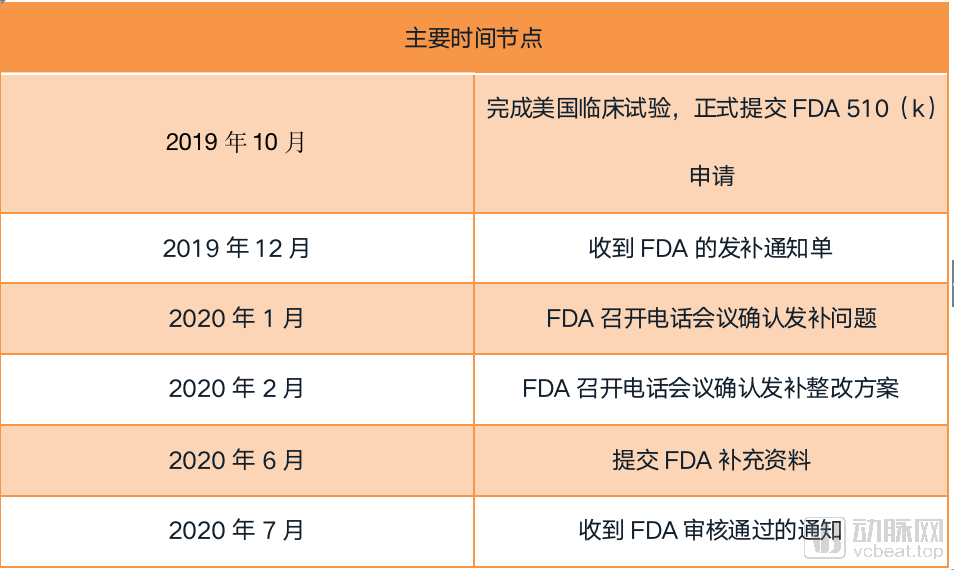
Looking back at the entire process, Infervision has summarized its experience in obtaining FDA approval into the following six points:
1. Building AI products that stand the test of time requires long-term technological accumulation and strong product development capabilities from excellent R&D personnel.
2. Build a professional certification team with international experience.
3. R&D is not an isolated function; it requires collaborative efforts across R&D, product, scientific research, and regulatory certification teams.
4. Conduct in-depth research on comparable lung AI products and FDA regulations, which requires the team to be familiar with the FDA’s regulatory requirements for similar products.
5. It is inferred that Infervision gained a first-mover advantage by proactively communicating with the FDA on key issues through pre-submission meetings.
6. Upon receipt of the FDA's request for additional information, Infervision will strive to clarify the specific requirements through the Q-sub (SIR) pathway as quickly as possible before submitting the response.
Summarizing the regulatory approval experience across various domains is not difficult, but putting it into practice is far from easy. Both our products and our team have been continuously refined over many years of Infervision’s deep engagement in medical AI, forming the company’s most formidable competitive moat.
As early as 2017, Infervision completed its international deployment in the United States, Japan, and Germany, and successfully deployed its AI-powered lung products in hospitals in Japan and Germany over the following year.
Three years later, Infervision’s international strategy has matured, pursuing a dual-track approach: one targeting developed countries such as Germany, the United States, and Japan; the other following the New Silk Road to deliver advanced medical AI technologies to countries in the Middle East and Africa.
Currently, Infervision’s overseas teams have covered ten countries through these two channels. Various teams are participating in international projects, advancing areas such as AI networks for the early diagnosis and treatment of cancer, AI systems for the prevention and control of infectious diseases, and AI infrastructure development.
Products targeting different countries inevitably face varying regulatory approval requirements, and Infervision has undoubtedly done extensive work in this regard.
On February 24, 2020, Infervision’s AI product, InferRead, obtained EU CE certification, becoming the first chest AI product certified in Europe. This achievement marked a perfect culmination of the Infervision team’s efforts since entering the European market in 2018.
In Europe, Infervision has similarly undergone a transition from skepticism to recognition, establishing robust trust and deep collaboration with partner hospitals.
Europe has a population of approximately 500 million. Based on U.S. rates, there are an estimated 14 million individuals eligible for early lung cancer screening, which would generate a lung cancer AI market worth billions of euros.
Meanwhile, Europe is placing increasing emphasis on early screening for lung cancer. Achieving early detection and treatment of lung cancer would generate substantial savings in healthcare costs for the entire medical system. Therefore, despite the standardized processes and high standards of care at most European hospitals, Infervision AI is still regarded as an indispensable clinical decision-support tool and a foundational platform for scientific research.
In June of the same year, Infervision obtained market access approval from Japan’s Pharmaceuticals and Medical Devices Agency (PMDA). Its AI-powered pneumonia product, developed in response to the COVID-19 pandemic, became the first COVID-19 AI-assisted diagnostic product approved by the PMDA, as well as the first lung-related AI-assisted diagnostic product approved by the agency.
These achievements collectively substantiate that Infervision’s FDA approval was no accident; it is the culmination of sustained long-term investment.
Looking Back at the First Half of 2020: The Chains That Have Constrained the Development of AI in Medical Imaging for Years Are Gradually Being Unlocked. In January, Keya Medical’s CT-FFR broke through the barriers, becoming the first AI company to receive Class III medical device approval from the National Medical Products Administration (NMPA). Lepu Medical followed closely, with its AI-based ECG analysis software obtaining a Class III certificate. In June, Ande Medintelligence’s MR AI product was approved, securing China’s first Class III certification for AI-based “image-assisted diagnosis” software. These achievements have reignited industry interest in medical AI. Now, with Infervision’s lung AI receiving FDA clearance, a beacon for the overseas commercialization of artificial intelligence is poised to be established.
However, the obstacles hindering the development of AI companies are not limited to regulatory reviews and approvals in various countries. As enterprises, they must consider scenario-specific needs and assess whether hospitals have the willingness to pay, thereby preventing the tragedy where many AI medical devices become obsolete immediately after approval.
Therefore, regardless of regulatory approval, market survival, or physician acceptance, the key factor remains the intrinsic value of artificial intelligence. Infervision has capitalized on North American demand; in their view, while regulatory approval is important, it is merely an interlude in product development—the ultimate goal is to identify clinical scenarios and achieve commercial implementation.
Moreover, the significance of Infervision’s FDA approval extends far beyond the healthcare industry. In fact, amid the many uncertainties characterizing current U.S.-China relations, this regulatory clearance reaffirms that medicine and technology know no borders.
This FDA certification also serves as a strong testament to the achievements attained by global medical AI enterprises. As Chen Kuan, Founder and CEO of Infervision, stated, “This certification carries profound historical significance. We firmly believe that even today, exchange and integration in people’s livelihood technologies between China and the United States remain both achievable and necessary, and there must be those who drive this progress. Infervision is but one such participant, yet we are unequivocally committed to forging ahead with determination.”