Tencent Launches iDrug, Its First AI-Powered Drug Discovery Platform, and Files for IPO

Tencent
Internet Comprehensive Service Provider
On July 9, the World Artificial Intelligence Conference (WAIC) Cloud Summit kicked off. At the event, Ren Yuxin, Chief Operating Officer of Tencent, announced the latest progress in leveraging AI to accelerate drug R&D: “iDrug,” Tencent’s first self-developed, AI-driven drug discovery platform, was officially released to the public.
The launch of the Yunshen Zhiyao platform will help researchers improve the efficiency of preclinical drug discovery, potentially alleviating the pharmaceutical industry’s urgent need for rapid, low-cost drug development amid the threat of the COVID-19 pandemic. Tencent has partnered with multiple pharmaceutical companies to apply AI models to real-world drug R&D projects. Currently, more than ten projects, including those related to the development of anti-SARS-CoV-2 drugs, are running stably on the Yunshen Zhiyao platform.
“Yunshen Zhiyao” draws its name from the Tang dynasty poem “Seeking the Hermit in Vain,” specifically the line “He is somewhere in this mountain, but lost in the depths of the clouds,” alluding to the similar journey underlying new drug development. The platform aims to cover the entire preclinical drug discovery process, comprising five core modules: protein structure prediction, virtual screening, molecular design/optimization, ADMET property prediction (to be open-sourced soon), and synthetic route planning.
Yunshen Zhiyao Platform: Functional Modules Cover the Entire Drug R&D Process
Protein structure prediction, as the foundation of drug design, is crucial for understanding molecular interactions within biological systems. Previously, pharmaceutical companies and research institutions relied on traditional experimental methods to determine protein structures, a process that was often difficult, time-consuming, and costly. By leveraging deep learning models to predict protein structures and functions, computers can rapidly and selectively identify potential hit compounds from hundreds of millions of small molecules, thereby significantly enhancing R&D efficiency.
On the YunShen ZhiYao platform, Tencent AI Lab has applied a new algorithm for predicting protein structures. Data show that Tencent’s new algorithm achieves a significant improvement on difficult cases (hard), outperforming Robetta, the industry-recognized authoritative method, by 10%.
Since joining CAMEO, the global authoritative benchmark for protein structure prediction, in 2020, the Tencent AI Lab team has secured the monthly championship five times within six months using its proprietary algorithm, outperforming numerous internationally renowned research teams and demonstrating robust technical capabilities. The innovative approach of this algorithm has also been integrated into the CloudDeep Intelligent Drug Discovery platform, where it will further deliver value in new target discovery and research into disease mechanisms.
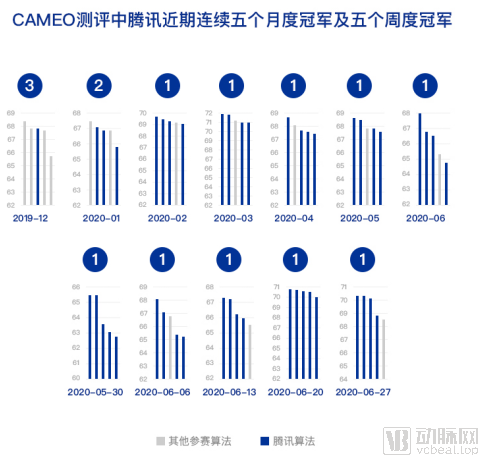
Vertical axis: lDDT, a score for protein structure prediction quality; a higher value indicates greater similarity between the predicted protein model and the actual protein structure.
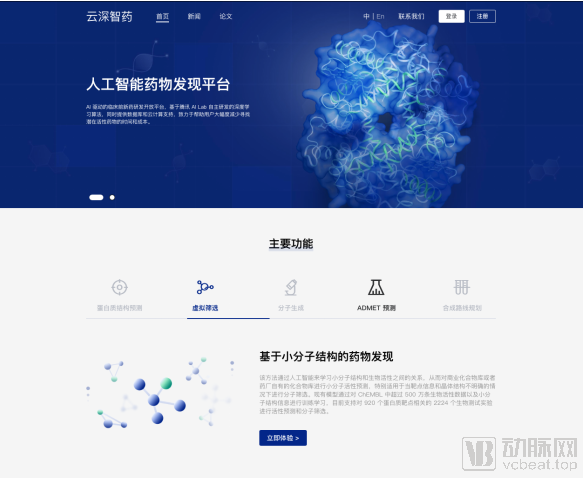
In the fields of virtual drug screening and ADMET property prediction, Tencent AI Lab has also achieved high accuracy on multiple public datasets, surpassing industry benchmarks. The ADMET prediction module will subsequently open-source the large-scale self-supervised molecular graph pre-training GX model, and the molecular generation model is expected to be open-sourced in the second half of the year.
Currently, the two tool modules for virtual screening and ADMET property prediction are available free of charge. Modules for protein structure prediction, molecular design/optimization, and synthesis route planning will be rolled out in the coming months, and the platform will continue to develop additional drug discovery and analytical function modules.
In addition to free access to the platform’s core features, pharmaceutical companies and research institutions can co-develop customized AI tools with Tencent. By integrating the strengths of Tencent AI Lab and Tencent Cloud in cutting-edge algorithms, optimized databases, and computing resources, the Yunshen Zhiyao (Cloud Deep Intelligent Drug Discovery) platform eliminates the need for users to deploy infrastructure themselves. Users can rapidly integrate AI capabilities into their existing R&D workflows simply by logging into the platform, thereby facilitating more efficient research.
As one of the key innovative technologies in the field of drug design, artificial intelligence (AI) and big data will bring new opportunities for intelligent transformation in drug research and development (R&D). Against the backdrop of China’s “New Infrastructure” initiative, Tencent will continue to deepen the integration of emerging technologies such as AI and big data with the needs of drug R&D. By leveraging advanced technologies to empower the industry and accelerate the high-speed development of China’s drug R&D sector, Tencent aims to provide robust technical support for innovation in the pharmaceutical industry.
AI Empowers Drug R&D: Algorithms, Computing Power, and Data Are Indispensable and Mutually ReinforcingAdvanced algorithms can deeply mine existing big data and analyze the implicit relationships within it. This process not only directly facilitates new drug discovery but also integrates vast amounts of existing databases, while promoting the generation and accumulation of new data to further optimize the algorithms. Optimized algorithms, in turn, reduce the model’s dependence on data volume and enhance its generalizability. Tencent’s robust computing power accelerates database storage and retrieval as well as algorithm iteration, significantly shortening the computational time required for model operations.
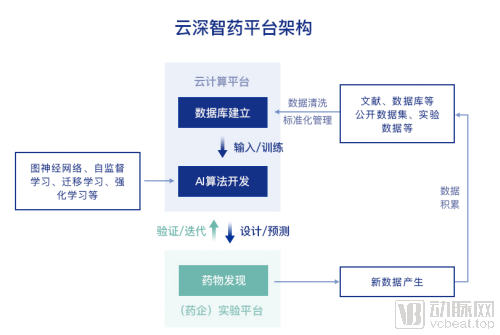
In addition to continuous innovation in algorithms, the Yunshen Intelligent Drug Discovery Platform also provides integrated support services for computing power and databases.
In terms of data, molecular big data serves as the infrastructure for drug discovery. Existing public datasets of drug molecules, represented by PubChem and ChEMBL, are derived from diverse sources. However, due to variations in experimental environments across different institutions, these datasets suffer from misalignment, extensive missing fields, and overall poor quality, making them difficult to use directly for developing predictive models. The molecular big data utilized by the DeepIntel Health platform is built upon existing public datasets and has undergone meticulous cleaning and curation across multiple stages. This process yields a comprehensive drug molecule dataset ready for direct construction of deep learning models. Its efficacy has been validated in numerous drug discovery projects, where the cleaning process significantly improved outcomes. The integrated large-scale dataset, resulting from the harmonization of multiple databases after rigorous cleaning, is being rolled out incrementally.
In terms of computing power, Tencent Cloud provides computational resources for the Yunshen Zhiyao platform. Pharmaceutical companies and research institutions can conduct research by simply logging into the platform, eliminating the need for self-deployment and enabling the rapid integration of AI capabilities into existing R&D processes.
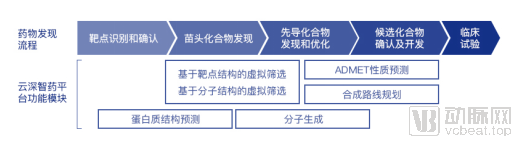
The preclinical new drug discovery process involves target identification and validation, hit discovery, lead discovery and optimization, and ultimately the confirmation and development of clinical candidate compounds. The “YunShen ZhiYao” platform covers the entire workflow of preclinical new drug discovery.
The first step in new drug discovery is target identification and validation, which involves locating the site of action of a drug within the body. Determining the structure of target proteins is a critical task in this process and is regarded as a fundamental cornerstone of drug development. For instance, if a protein is involved in a certain disease and serves as a key component in a critical pathway, researchers can design drug molecules to modulate the protein’s function once its structure is elucidated. Experimental determination of protein structures is often challenging, time-consuming, and costly. By leveraging deep learning models to predict protein structures and functions, computers can more rapidly and selectively identify potential hit compounds from hundreds of millions of small molecules.
The protein structure prediction method adopted by the “Yunshen Zhiyao” platform has achieved international leading levels of accuracy, thanks to breakthroughs in two key technologies. First is a protein folding method based on self-supervised learning that does not rely on homologous sequences; instead, it learns co-evolutionary patterns directly from sequence databases through self-supervised learning, thereby generating pseudo-homologous sequences containing co-evolutionary information de novo, which ultimately enables effective folding of these proteins. Second is an iterative deep learning-based approach that effectively integrates template-based modeling with free modeling, proposing for the first time dynamic, iterative, amino acid pair-specific constraints, significantly improving modeling accuracy and thus enabling better protein folding.
Hit compound screening against targets is the second step in new drug discovery. Compared with traditional experimental screening, virtual screening performed via computational methods does not consume compound samples, thereby significantly saving manpower and material resources. Ligand-based drug design (LBDD) is one of the common methods for virtual screening. It involves learning and establishing models of the relationship between molecular structure and activity based on the structures of known active small-molecule ligands, to predict the activity of new compounds. However, the limited availability of measured compound activity data for many targets severely constrains the accuracy of predictive models. AI methods are expected to address this issue. For instance, the virtual screening module of the “Insilico Medicine” platform has, for the first time, applied meta-learning and deep neural network algorithms to LBDD tasks. By leveraging AI-driven “transfer” of knowledge learned from other targets (such as the impact of local molecular structures on target binding affinity) to the target of interest, the platform improves model prediction accuracy. Currently, this algorithm has increased the median prediction accuracy (correlation between predicted and experimentally measured activities) from the previous record high of 0.36 to 0.42 across thousands of experimental datasets, while raising the percentage of viable screening models from 56% to 60%, surpassing industry standards.
In the late stages of drug development, predicting the ADMET properties of molecules—encompassing absorption, distribution, metabolism, excretion, and toxicity—is particularly critical. Statistics indicate that up to 60% of late-stage drug failures are attributable to ADMET-related issues. Therefore, early identification and elimination of molecules with poor druggability can significantly reduce the risk of failure in later phases of drug development. AI-based ADMET prediction enables medicinal chemists to rapidly modify molecular structures, optimize physicochemical properties, shorten drug development cycles, and lower experimental testing costs. The small-molecule ADMET property prediction module of the "Yunshen Zhiyao" platform has outperformed the best existing academic models by 3%–11% across multiple datasets. According to feedback from partners, the platform’s proprietary algorithms exceed the accuracy of current commercial software by 6%–37%. Furthermore, the platform employs mechanisms such as attention visualization to illustrate the impact of molecular substructures on prediction results, thereby enhancing model interpretability. Additionally, the platform offers flexible deployment options, including on-premises versions, to ensure user data security.