Inozyme Pharma Raises $128.8M in IPO to Advance Novel Therapies for Rare Abnormal Mineralization Disorders

Inozyme Pharma
Rare Disease Biopharmaceutical Manufacturer
Recently, VCBeat learned thatU.S. biopharmaceutical company Inozyme Pharma, listed on the Nasdaq under the ticker symbol “INZY,” conducted an initial public offering of 7,000,000 shares of common stock at a price of $16.00 per share on its listing day.As of July 30, following the exercise of the over-allotment option, Inozyme Pharma issued a total of 8.05 million shares, with the final offering size reaching $128.8 million (approximately RMB 895 million).
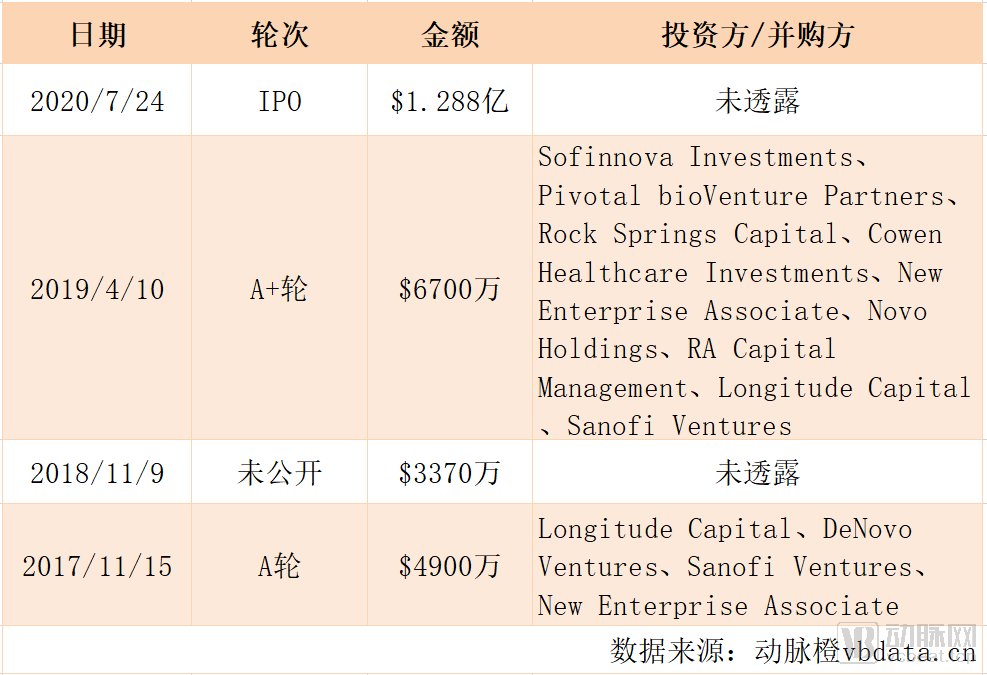
Inozyme Pharma's Historical Financing
Inozyme Pharma, founded in 2016 and headquartered in Boston, Massachusetts, is a biopharmaceutical company focused on rare diseases. It is dedicated to developing novel therapies for disorders of abnormal mineralization, primarily ENPP1 deficiency and ABCC6 deficiency. These conditions affect the blood vessels, soft tissues, and bones, leading to progressive debilitation and potentially life-threatening complications.
Axel Bolte is the President, Chief Executive Officer, and Co-Founder of Inozyme Pharma, and a member of the Board of Directors of IVERIC bio. Mr. Bolte earned his undergraduate degree in Biochemistry from ETH Zurich (Swiss Federal Institute of Technology) in Zurich, Switzerland, and later obtained a Master of Business Administration (MBA) from the University of St. Gallen in Switzerland.
Prior to joining Inozyme, Bolte served as a Venture Partner and Investment Advisor at HBM Partners AG, an investment advisory services provider specializing in the life sciences industry, and spent 16 years working in healthcare venture capital. Before founding Inozyme, Bolte focused extensively on the healthcare sector, particularly in pharmaceuticals, biotechnology, and drug development. He has accumulated extensive experience in pharmaceutical deal sourcing, transaction structuring, and joint monitoring. Bolte was also the founder of Turos Capital AG and served as a board member for Allena Pharmaceuticals, Nabriva Therapeutics, and PTC Therapeutics.
Abnormal mineralization diseases, a class of inherited rare disorders, are caused by pathological mutations in patient-related genes that lead to metabolic abnormalities in the body's mineralization pathways.
In a properly functioning mineralization pathway, the enzyme encoded by the ENPP1 gene cleaves ATP into pyrophosphate (PPi) and adenosine monophosphate (AMP). PPi is a potent regulator of mineralization that controls the rate of calcium crystal deposition in bone. Meanwhile, AMP is further metabolized into adenosine, a powerful modulator of cell proliferation that plays a significant role in regulating vascular responses to injury, while also preventing neointimal hyperplasia and excessive growth of vascular smooth muscle cells.
ENPP1 DeficiencyIt is a rare congenital genetic disorder caused by metabolic errors due to ENPP1 gene mutations, commonly referred to in medical literature as Autosomal Recessive Hypophosphatemia Type 2 (ARHR2). Approximately 11,000–12,000 individuals worldwide are affected by this condition. As an autosomal recessive disorder, it results from genetic mutations that lead to reduced or absent ENPP1 enzyme activity.
ENPP1 deficiency leads to low levels of pyrophosphate (PPi) and AMP (a precursor of adenosine) in plasma, which can result in vascular neointimal hyperplasia, accompanied by higher early mortality and long-term morbidity. ENPP1 deficiency occurs throughout a patient's life, beginning as early as fetal development and continuing into adulthood.
Children with ENPP1 deficiency are characterized by persistent vascular and organ calcification, as well as the development of rickets. Persistent arterial calcification in children stimulates the production of fibroblast growth factor 23 (FGF23), a hormone that causes renal complications and contributes to disease progression. This condition leads to severe skeletal deformities, resulting in short stature, severe bone pain, and an increased risk of fractures. Furthermore, children with ENPP1 deficiency may experience excessive joint calcification and dental problems due to malformation of permanent teeth, significantly impairing their quality of life.
In adulthood, ENPP1 deficiency manifests as osteomalacia, in addition to ongoing vascular and organ calcification, leading to severe bone pain, fatigue, muscle weakness, and an increased risk of fractures. Adults with ENPP1 deficiency experience significant functional and cognitive impairments, severely limited ability to perform activities of daily living, and reduced quality of life.
ABCC6 DeficiencyIt is a rare congenital genetic disorder caused by mutations in the ABCC6 gene, leading to metabolic errors. This disease affects more than 67,000 people worldwide. ABCC6 deficiency is also an autosomal recessive disorder, where genetic mutations result in reduced or absent activity of the ABCC6 protein. ABCC6 deficiency leads to decreased levels of pyrophosphate (PPi) in plasma, which in turn causes pathological mineralization of systemic blood vessels and soft tissues throughout the body. ABCC6 deficiency can lead to blindness, life-threatening cardiovascular complications, and skin calcification in patients.
Abnormal mineralization and neointimal hyperplasia may also occur in non-genetic diseases.
Neointimal hyperplasia in the vascular system is a hallmark of many non-genetic disorders, including calciphylaxis. Calcification inhibition is a manifestation of chronic kidney disease associated with low levels of pyrophosphate (PPi). It is characterized by pathological calcification of the vasculature in the skin and adipose tissue, which subsequently leads to thrombosis and cutaneous ulcers.
Although the etiology of this type of abnormal mineralization disease differs from that of genetic abnormal mineralization diseases, both are caused by abnormal levels of pyrophosphate (PPi) in the patient’s body, which has enabled Inozyme to expand the indications for its drug.
In light of this area of high unmet medical need, Inozyme has launched the INZ-701 research program.INZ-701 is a soluble protein designed to correct mineralization pathway defects caused by mutations in the ENPP1 and ABCC6 genes.As mentioned above, this mineralization pathway is key to regulating calcium deposition in the human body and is also associated with neointimal hyperplasia and excessive growth of vascular smooth muscle cells.
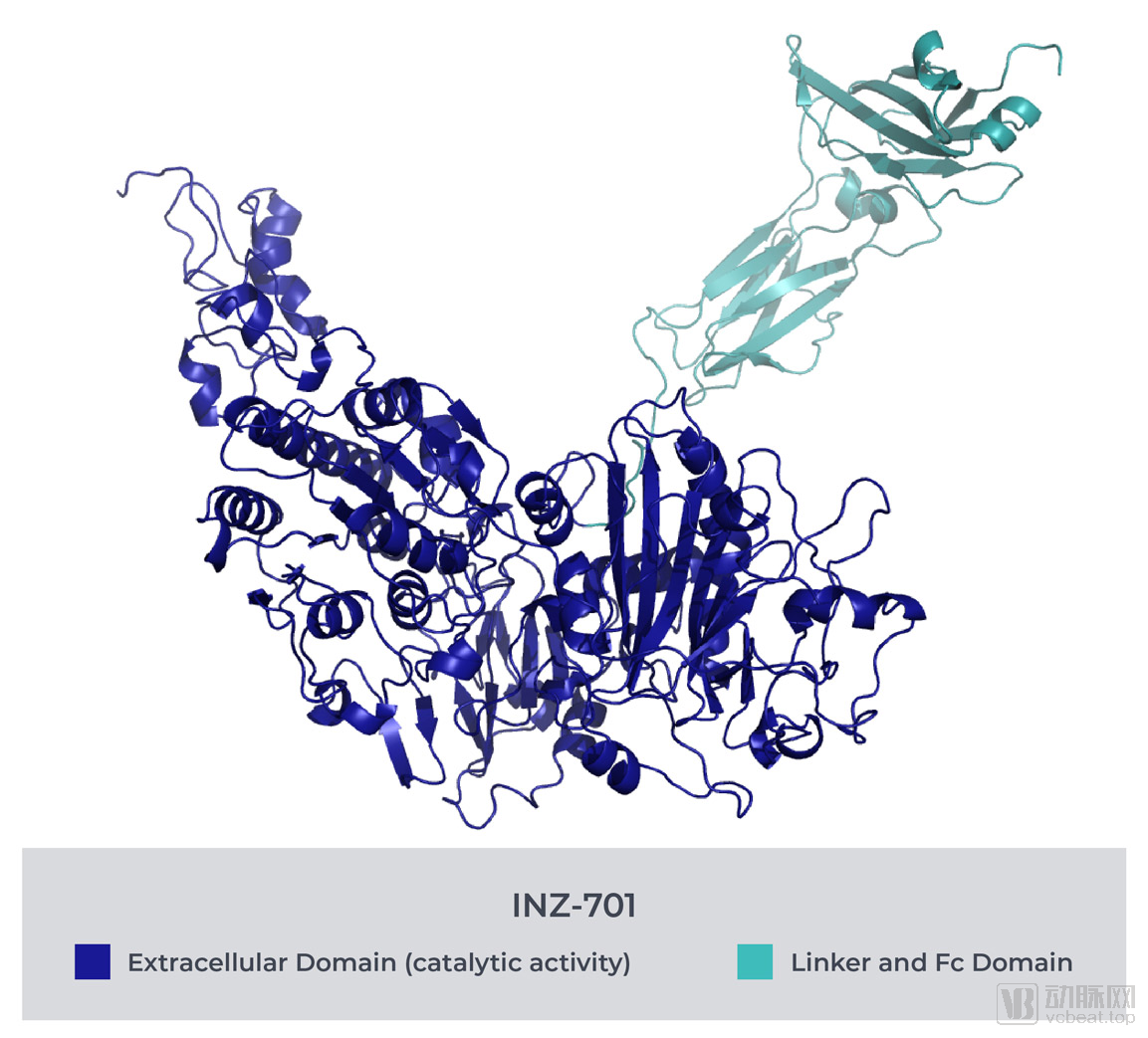
INZ-701 restores the normal balance of pyrophosphate (PPi) and adenosine in ENPP1 deficiency, compensating for the loss of enzymatic function caused by monogenic mutations in ENPP1 or ABCC6. This helps maintain homeostasis and ensures that related metabolic processes proceed normally.
INZ-701 is administered via subcutaneous injection. Following administration, INZ-701 maintains stable blood drug concentrations, with therapeutic effects typically lasting up to one week, thereby eliminating the inconvenience of frequent dosing intervals.
INZ-701 aims to “mimic” the role of native ENPP1. Like native ENPP1, INZ-701 catalyzes the metabolism of extracellular ATP and other nucleotides, cleaving ATP into PPi and AMP (a precursor of adenosine).
Currently, Inozyme’s INZ-701 program has successfully completed preclinical animal studies, yielding robust data to support subsequent development. The program is also protected by proprietary intellectual property rights.
In recent years, the U.S. government has introduced a series of policies to intensify efforts in rare disease drug development and encourage pharmaceutical companies to research treatments for rare diseases. This has led many enterprises to view the United States as an incubator for developing “blockbuster” orphan drugs. Measures such as shortening the approval cycle for rare disease drugs and granting priority review privileges have prompted numerous pharmaceutical companies to join the surge in rare disease drug development. Inozyme is undoubtedly one of them.
Currently, INZ-701 has been granted orphan drug designation by the U.S. Food and Drug Administration (FDA) and the European Medicines Agency (EMA) for the treatment of ENPP1 deficiency.This means that INZ-701 will be eligible for a 50% tax credit and waiver of user fees, and will receive seven years of market exclusivity upon successful commercialization.
Inozyme Pharma expects to submit an Investigational New Drug (IND) application to the U.S. Food and Drug Administration (FDA) and a Clinical Trial Application (CTA) in Europe for INZ-701 in the second half of this year, after which it will advance the INZ-701 program by initiating two separate Phase 1/2 clinical trials. One trial will be conducted in the United States and Europe, specifically targeting patients with ENPP1 deficiency; the other will be conducted in Europe, specifically targeting patients with ABCC6 deficiency.