Celebrity Surrogacy Scandal Puts China's ART Industry Under Spotlight Amid Regulatory Tightening and Market Growth
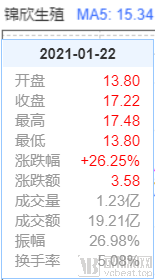
In recent days, the surrogacy scandal involving a Chinese celebrity has sparked widespread public outcry. Amidst the fallout, Jinxin Fertility, China’s leading listed company in assisted reproductive technology, saw its stock price surge on January 18, the day the news broke, continuing to rise for three consecutive days. As of January 22, one week after the incident emerged, Jinxin Fertility’s weekly gain had exceeded 26%.
Surrogacy Is Not Equivalent to Assisted Reproductive Technology (ART), assisted reproductive technology (ART) is merely the technical means required for surrogacy. With the support of ART, after a couple completes in vitro fertilization (IVF), the resulting embryo is typically transferred back into the uterus of the woman who provided the oocyte, where it develops normally to term. However, if the embryo is transferred into the uterus of a third-party woman for gestation, this constitutes surrogacy. From a technical perspective, human surrogacy is fully feasible; however, from legal and ethical standpoints, surrogacy is unequivocally illegal and unrecognized in China.
As early as 2001, the “Administrative Measures for Human Assisted Reproductive Technology” promulgated by the former Ministry of Health, together with the “Ethical Principles for Human Assisted Reproductive Technology and Human Sperm Banks” issued in 2003, explicitly stated thatMedical institutions and medical personnel are prohibited from implementing any form of surrogacy technology.. The prohibition of surrogacy not only protects the interests of vulnerable women and children, but also serves as a wake-up call for the standardized development of the assisted reproductive technology industry.
The assisted reproductive technology (ART) industry in China is a specialized healthcare vertical whose marketization and maturity are steadily improving, encompassing multiple subsectors including therapeutic services, medical consumables, biopharmaceuticals, and genetic testing. Unlike other healthcare verticals, the pharmaceutical sector focuses on human longevity, whereas ART centers on human fertility and reproduction. Fertility has long been a global concern, and the underlying demographic composition even foreshadows a nation’s future development.
In recent years, China has continuously relaxed its fertility policies, from allowing two children to permitting three. This is a response by the government to the accelerating trend of population aging in society. Although the state has provided comprehensive policy support for childbirth, increasing life pressures on citizens in modern society, along with negative impacts from living environments and dietary health, have made "unwillingness to have children" and "inability to have children" two major obstacles standing in the way of fully liberalizing childbirth. Addressing "unwillingness to have children" requires reforms in various supporting systems and policies related to childbirth at the national level, while tackling the core reason behind "inability to have children"—infertility—relies on advancements in assisted reproductive technologies. In other words,In China, assisted reproductive technology is primarily used to address infertility issues in couples, rather than serving as a tool for surrogacy or embryo sex selection.。
We provide a brief overview of China’s recent regulatory developments in the assisted reproductive technology (ART) industry and, by drawing on experiences from Europe and the United States, offer insights into the future trajectory of ART development in China.
1. Macro-Policy Boundaries of China’s Assisted Reproductive Technology Industry
2. Accelerated Approval of Clinical Exemptions for Upstream Devices in Assisted Reproductive Technology
3. Reference Value of European and American ART Industry Regulation for China
China’s systematic regulation of the assisted reproductive technology (ART) industry began with the promulgation of the first Administrative Measures for Human Assisted Reproductive Technologies in 2001. In these Administrative Measures, Chapter I, General Provisions, emphasizes “The buying and selling of gametes, zygotes, and embryos in any form is prohibited. Medical institutions and medical personnel shall not implement any form of surrogacy technology.”, which established the fundamental implementation boundaries for assisted reproductive technologies in China.

Regulatory Documents on Assisted Reproductive Technology Issued in China
These Administrative Measures came into effect on August 1, 2001. They set forth the conditions that medical institutions must meet to provide assisted reproductive technologies (ART), outlined the approval procedures, and provided systematic guidance on implementation processes and penalties for violations. Concurrently, the Administrative Measures for Human Sperm Banks were promulgated to support the safe, effective, and healthy development of human assisted reproductive technologies.
"Administrative Measures for Human Sperm Banks" formulates plans for the establishment of human sperm banks based on actual conditions in China, including health resources, demand for donor sperm, sperm sources, and technical requirements. The approval certificate for a human sperm bank is subject to validation every two years. Sperm donors must be healthy males aged 22 to 45 years and may donate sperm to only one human sperm bank.No more than five women shall be provided with conception services.and other specific instructions.
The “Administrative Measures for Human Assisted Reproductive Technology” and the “Administrative Measures for Human Sperm Banks” are the “two major measures” that establish the foundational framework for assisted reproduction in China. Following the promulgation of these two measures, the corresponding “two major technical specifications” were subsequently issued: the “Technical Specifications for Human Assisted Reproductive Technology” and the “Basic Standards and Technical Specifications for Human Sperm Banks.”
The “Two Major Technical Specifications” issued by the National Health Commission’s Department of Science, Education and Technology in 2001 underwent one revision in 2003. The final 2003 revised version of the Basic Standards and Technical Specifications for Human Sperm Banks set forth explicit requirements for the establishment conditions, staffed personnel, facilities, and equipment of institutions providing assisted reproductive technologies. It also stipulated that no reproductive institution may perform more than 1,000 oocyte retrieval cycles per year of in vitro fertilization and embryo transfer and their derived technologies, andStrictly prohibit pregnancy and childbirth for third child and above。
Meanwhile, the technical specification explicitly states: implementing technical personnelSex selection without medical indications is prohibited; the implementation of surrogacy techniques is prohibited; the donation of embryos is prohibited; genetic manipulation of human embryos for reproductive purposes is prohibited; hybridization between human and non-human gametes is prohibited; research on human chimeric embryos is prohibited; and human cloning is prohibited.The 15 prohibitive regulations have drawn clear boundaries for the practice of assisted reproductive technology.
Meanwhile, the "Basic Standards and Technical Specifications for Human Sperm Banks" further specified the establishment conditions and management requirements for human sperm banks. In the same year, the state also promulgated the "Ethical Principles for Human Assisted Reproductive Technology and Human Sperm Banks" from an ethical perspective, laying the theoretical foundation for the development of assisted reproduction.
Following the issuance of a series of macro-level regulatory documents on assisted reproduction by the state, China’s assisted reproduction industry has been broadly segmented into upstream medical enterprises supplying devices, consumables, diagnostic technologies/reagents, and biopharmaceuticals, and downstream medical institutions providing assisted reproduction services. According to statistics, currently in ChinaA total of 517 medical institutions are approved to provide human assisted reproductive technology services., with a total of 27 medical institutions approved to establish human sperm banks, distributed across major provinces and municipalities.
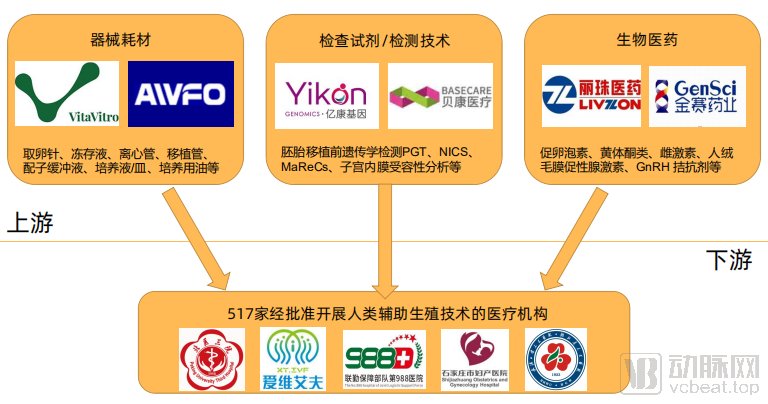
Domestic Upstream and Downstream Layout of Assisted Reproductive Technology
The approval process for upstream ART-related biopharmaceuticals and conventional new drugs is consistent, so it will not be elaborated here. We primarily focus on the regulatory approval of upstream ART medical devices and consumables, as well as the approval and management of genetic testing reagents.
Medical devices and diagnostic reagents both fall under the NMPA’s medical device approval category. However, their approval process differs from that of pharmaceuticals, which has largely become systematic and standardized (preclinical → IND → Phase I, II, and III clinical trials → NDA). Due to the wide variability among medical device products, they are classified into Class I, II, and III medical devices in descending order of usage safety risk. Class I medical devices are exempt from clinical trials, while Class II and III medical devices are primarily evaluated based on whether they are included in the list published by the NMPA."Catalogue of Medical Devices Exempt from Clinical Trials"(hereinafter referred to as the "Exemption from Clinical Trials Catalog") to determine whether clinical trials are required. However, the clinical trial design for medical devices is often relatively straightforward, primarily aimed at supporting pre-market registration.
According to the "Medical Device Classification Catalog" (No. 104 of 2017), the classification code for assisted reproductive technology devices is 18-07, whereinLiquid products and laser systems for assisted reproduction are classified as Class III; all others are Class II.. It is worth noting that, in essence, assisted reproductive testing reagents are IVD kits based on gene sequencing, and they are still classified as Class III medical devices by the state.
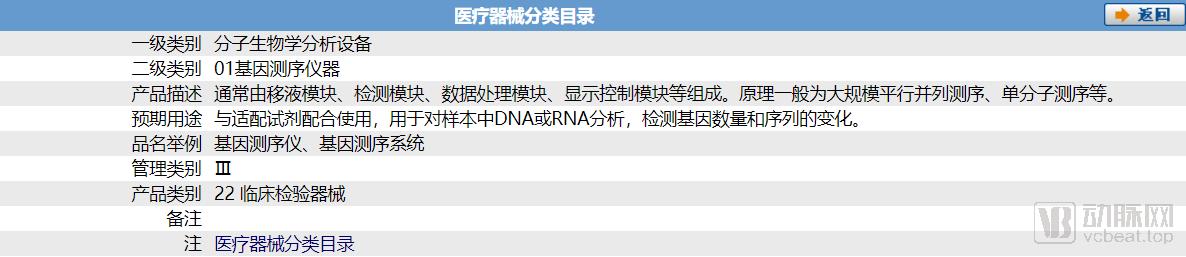
ART-related genetic testing kits have been included in the national Class III medical device catalog
And in February 2020,Berry Genomics' PGT-A Kit(Preimplantation Chromosomal Aneuploidy Testing Kit) was the first to obtain Class III medical device certification from the NMPA (National Medical Products Administration Registration No.: 20203400181),Becoming the first in China to obtain approval for ART genetic testing reagentsmedical company. The approval of the kit facilitates the large-scale application of preimplantation genetic testing for aneuploidy (PGT-A) in assisted reproductive technology (ART). In the same year, the General Office of the National Medical Products Administration issued Announcement No. 60 of 2019, which published 24 medical device industry standards, including YY/T 0506.8—2019 “Surgical drapes, gowns and clean air suits for patients, clinical staff and equipment – Part 8: Particular requirements for products,” thereby incorporating PGT-A kits into the national mandatory pharmaceutical industry standards.
Led by PGT-A kits, it is foreseeable that future ART-related genetic tests, including PGS and PGT-SR, will be further standardized and incorporated into the national approval and regulatory framework for Class III medical devices.
Notably, upstream suppliers of genetic testing solutions for assisted reproductive technology (ART), in addition to offering commercial channels for in vitro diagnostic (IVD) kits, can now empower downstream ART centers through third-party clinical laboratory services. For instance, Yikang Gene’s strategic presence in the ART sector provides comprehensive coverage of genetic testing across the entire ART workflow. The genetic testing needs of ART centers can thus be met by collaborating with third-party clinical laboratories such as Yikang Gene.
Between 2001 and 2003, China formulated administrative measures, technical specifications, basic standards, and ethical principles related to assisted reproductive technology (ART) and human sperm banks, which have since served as guiding standards for the development of the ART industry. Meanwhile, the National Medical Products Administration (NMPA) issued a series of guidelines for medical device registration and approval, further standardizing the clinical application of ART. Since 2015, specialized regulatory documents and standards pertaining to ART-related medical devices have been progressively introduced. To date, the following guideline documents and industry standards have been published:

Regulatory Documents on Assisted Reproductive Technology Devices Issued by the National Medical Products Administration
It can thus be seen that the technical review guidelines and standards currently issued in China do not cover all assisted reproductive medical device products, particularly industry standards, which only encompass two Class II products (Embryo Transfer CatheterandOocyte Retrieval Needle for Assisted Reproduction) have clear industry standard requirements. As mentioned above, domestic manufacturers of assisted reproductive technology (ART) devices started later than their foreign counterparts, with lower product maturity and smaller enterprise scale compared to many imported brands. Furthermore, strategies for domestic registration submissions are still in their infancy. Therefore, the registration and approval process for ART products still requires further exploration and refinement.
Meanwhile, from the perspective of clinical evaluation pathways, Class II medical devices have largely been included in the list of devices exempt from clinical trials, and a significant proportion of Class III devices have also been added to this list. This has greatly facilitated the registration of such products, not only saving costs and resources associated with clinical trials but also reducing the time required for clinical validation during the design and development phase for manufacturers, thereby accelerating the market launch of new products.
“Previously, due to the stringent registration requirements in China, where nearly all assisted reproductive technology (ART) products required clinical trials, some technologies and products still in use—or to which healthcare professionals had become accustomed—remained from the previous generation,” an ART industry insider told VCBeat. “However, by the end of 2019, the national regulatory authorities exempted certain ART products from mandatory clinical trials for registration, accelerating the registration process for companies and benefiting both imported and domestically produced brands.”
Currently, there are medical device registration certificates for some domestically produced Class II ART products in China. However, it is extremely rare for domestically produced Class III liquid products to obtain domestic approval, and most of them were approved after being exempted from clinical trials. The original market for these products was basically dominated by imported products, and it is urgent for domestic companies to catch up. Fortunately,Chengdu AviveSelf-developedOil for Assisted Reproductive CultureIssued by the NMPA on August 18, 2020Class III Medical Device Registration Certificate, becomingChina's First Approved Culture Oil for Assisted ReproductionCoincidentally, also on January 22, 2021,Weituo BioSelf-developedVitrification Cryoprotectant Solutionalso obtained the certification issued by the NMPAClass III Medical Device Registration Certificate, becomingChina's First Approved Vitrification Cryopreservation Solution。

It is foreseeable that,With the national exemption of clinical trials for certain assisted reproductive technology (ART) devices, the approval and market launch timeline for ART-related devices and consumables in China will be significantly accelerated.However, it is worth noting that clinical requirements for medical devices are less stringent than those for pharmaceuticals. This may make it difficult for some medical device companies to voluntarily conduct various forms of spontaneous trials when not mandated by regulations. Although their products have already been launched on the market, they still face a new barrier when competing for market share against imported medical devices.
This may require not only market education to foster recognition of domestic brands, but also strict self-discipline and a commitment to excellence in independent research and development by medical device manufacturers.Lin Xiaozhen, Founder of Weituo BioIt is emphasized that although the National Medical Products Administration does not mandate clinical trials for the approval of the company’s vitrification freezing solution—allowing registration and market launch upon completion of a comparative analysis of clinical trial data from equivalent medical devices—the company, adhering to a rigorous approach, has independently conducted and completed clinical trials in China.Yan Fei, Founder of Chengdu iVfThe company also told VCBeat that its independently developed full-process embryo culture medium received China’s first clinical trial approval in 2020. It believes that for products guided by the value of meeting genuine clinical needs, gaining clinical recognition is only a matter of time.
Medical device standards are technical regulations that must be jointly observed in the research and development, manufacturing, distribution, use, and regulatory supervision of medical devices. As a technical foundation for regulation, they serve as the basis for registration testing and technical review, as well as for assessing compliance with quality management systems in production and imposing supervisory penalties. Medical device standards are also closely linked to industry development; they can standardize manufacturing and testing processes, reduce costs, and improve efficiency. Conversely, poorly formulated standards may hinder industrial growth and even cause disorder.
The U.S. Food and Drug Administration (FDA) was the earliest agency to focus on and implement safety evaluations and regulatory oversight for medical devices related to assisted reproductive technology (ART). Item G of Part 884 in Title 21 of the Code of Federal Regulations (CFR) specifies the classification and definitions of human ART-related medical devices. Under this classification framework, most such products are categorized as Class II medical devices (subject to special controls) and managed through the 510(k) premarket notification pathway involving substantial equivalence comparisons. This approach outlines overarching technical requirements for different product categories, including mouse embryo assays, endotoxin testing, sterilization validation, design performance specifications, biocompatibility testing, labeling requirements, and clinical testing.
Under U.S. regulations, ART-related medical devices are classified under obstetrics and gynecology devices and are regulated separately from other devices such as contraceptives.

Regulatory Frameworks for ART Devices in Europe and the United States
In Europe, the European Union is currently implementingMedical Devices Directive (MDD 93/42/EEC)Management of Non-active Medical Devices.
In 2017, the Official Journal of the European Union officially published the European Union Medical Device Regulation (hereinafter referred to as “MDR”). The MDR entered into force on May 26, 2017, and was originally scheduled to replace the old Medical Devices Directive (MDD) after a three-year transition period on May 26, 2020. However, due to the impact of the COVID-19 pandemic, the EU had to announce a one-year postponement of the MDR’s application date to May 26, 2021. Prior to this date, passive medical devices remained regulated under the MDD.
Beyond the MDD or MDR, the European Union issued the "Guideline on the Conformity Assessment of In Vitro Fertilization and Assisted Reproductive Technology Products" in 2012 to provide guidance for the compliance of medical devices used in assisted reproduction. This guideline covers medical devices related to IVF and ART listed in Annex IX of Directive 93/42/EEC, managing ART-related medical devices from a risk management perspective. The guideline emphasizes that the risks and hazards associated with IVF/ART products are linked to their design and manufacturing processes.
EU regulations (including the MDD and MDR) emphasize that the assessment of adverse effects of medical devices and the evaluation of an acceptable risk/benefit ratio mustBased on comprehensive preclinical evaluation and clinical data assessment, with particular emphasis on post-market clinical follow-up. Since adverse events associated with assisted reproductive technology do not necessarily occur immediately after the procedure but may arise after fetal birth or even later, the European UnionEmphasizes the traceability of vigilance and clinical evaluation for medical devices。
Currently, the MDR has not been formally implemented. Although some notified bodies are only accepting certification applications under the MDR, the EU has yet to issue relevant Product Specifications (PS). Consequently, strategies and processes for CE certification remain immature, particularly for assisted reproductive technology devices containing special components.
In summary, currentlyThe United States Has the Most Mature Regulatory Framework for Assisted Reproductive Technology Products, they do not classify such products under the highest level of regulatory oversight; market access requires only the standard premarket notification 510(k); secondly,The EU’s new Medical Device Regulation (MDR) imposes the strictest oversight on assisted reproductive technology products.Any device that comes into direct contact with gametes or embryos is classified as Class III. This means that, in addition to liquid products, passive devices such as needles, catheters, and culture dishes that come into contact with gametes or embryos—which are classified as Class II in China—will be classified as Class III under the MDR.
In comparison, China’s regulatory requirements for ART devices fall between those of the United States and the European Union. Although the pre-market approval process is not as streamlined as in the U.S., it is less complex than in the EU for most product categories. A significant proportion of devices are classified as Class II, and a considerable number of Class III devices have been included in the list exempt from clinical trials. It is believed that as the state further refines its regulatory framework for the ART industry, it will inevitably foster the growth of domestically produced ART brands. Moreover, with the intensifying national emphasis on encouraging childbirth, the assisted reproductive technology sector is poised to become an emerging high-growth industry in the future.
Special Thanks:
Yan Fei, Founder of iVf
Lin Xiaozhen, Founder of Weituo Bio
Data support from Hezhuo Biotech, Beikang Medical, and Yikang Gene