Is Gene Sequencing Accurate? A Technical Explanation from First Principles
A gene is a segment of DNA that carries genetic information. Through transcription and translation, it directs protein synthesis to express the genetic information carried by an individual, thereby controlling the expression of biological traits.
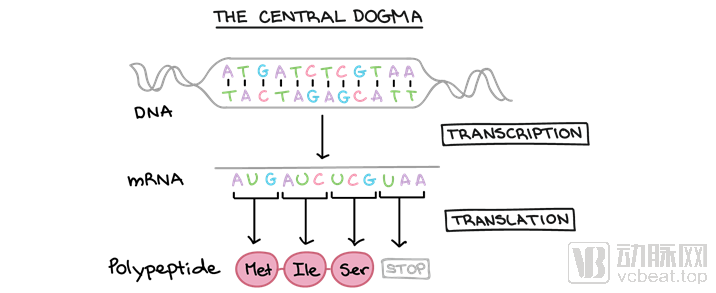
The Central Dogma: This refers to the process by which genetic information is transcribed from DNA into RNA and then translated from RNA into protein, thereby completing the transcription and translation of genetic information. (Image source: Khan Academy)
Genetic testing is a technique that analyzes DNA from blood, body fluids, or cells. It uses specialized equipment to examine the DNA molecular information within an individual’s cells, thereby identifying gene types and genetic defects and assessing whether their expression functions are normal. Genetic testing is a specialized diagnostic approach that integrates multiple disciplines, including molecular biology, laboratory medicine, and bioinformatics.
How to Demonstrate the Accuracy of Genetic Testing: The Industry Seems to Lack a Clear and Accessible Explanation. Drawing on the author’s clinical practice, this article aims to share insights with readers, who may include both general users interested in genetic testing and professionals. We welcome further discussion and exchange among peers to jointly promote the development of the genetic testing industry.
1"So-called 'Test Accuracy'"
When questions such as “Are the test results accurate?” are raised, concerns generally focus on two aspects: first, the accuracy of the measured data; and second, the accuracy of clinical interpretations regarding an individual’s health status (e.g., presence or absence of disease) based on those results. The former is a matter of metrology, while the latter pertains to clinical application. Ultimately, the clinical application of laboratory results is the primary objective.
Generally, the public’s understanding of “test accuracy” refers to the initial concern about whether the test results are accurate when receiving a laboratory report, particularly when abnormal results are reported. In clinical practice, this typically involves determining whether test indicators fall within the normal range (e.g., red blood cell count) or whether test results are positive or negative (e.g., hepatitis B virus antigen and antibody tests). Such results are directly interpretable and represent clinically validated indicators established through extensive prior experience. Therefore, it can be stated that, in the vast majority of cases, these conclusions align with clinical manifestations.
However, such evaluation criteria cannot be applied to the field of genomic variation and disease association. This is an emerging field, representing humanity’s first steps in exploring the unknown—a prolonged process akin to crossing a river by feeling for stones in the dark. During this crossing, some stones are clearly identified, others are less certain, some have been misidentified, and in some areas, it remains unclear whether any stones exist at all. Therefore, defining whether our next-generation sequencing-based analysis of rare genetic diseases is “accurate” cannot be generalized; instead, it requires a detailed and systematic review.
Therefore, it is essential to elaborate on the principles of sequencing, disease databases, and the development and technological advancements in bioinformatics analysis.
2Sequencing Principles and Sequencing Accuracy
DNA Sequencing Technology: The Technique for Determining the Order of DNA Bases. The human genome is composed of four deoxyribonucleic acids—adenine (A), guanine (G), thymine (T), and cytosine (C)—arranged in various sequences. Determining their sequence enables further research or modification of target fragments. Based on chronological emergence and differing sequencing strategies, current sequencing technologies are broadly categorized into first-generation sequencing (Sanger sequencing), second-generation sequencing (Illumina, Thermo Fisher, BGI, etc.), and third-generation sequencing (Oxford Nanopore Technologies, PacBio, etc.).

Evolution of Sequencers Across Generations: First-generation sequencing is unequivocally represented by ABI’s (now Thermo Fisher Scientific) Sanger sequencing. Second-generation sequencing originated with Roche 454 and now includes brands such as Illumina, Thermo Fisher Scientific (Ion Torrent), and BGI. Third-generation sequencing is currently dominated by PacBio and Oxford Nanopore Technologies (ONT). However, two Chinese sequencing companies—Qitan Technology and Jinshi Technology—have recently developed third-generation nanopore sequencers.
Although we commonly use the terms first-, second-, and third-generation to distinguish these sequencing technologies, they can be further subdivided within the industry. For instance, Illumina’s technology is referred to as high-throughput sequencing based on cluster generation, while Oxford Nanopore Technologies (ONT) is known as nanopore sequencing. In reality, none of these three generations of technology is inherently superior to the others, nor can any single technology currently replace the rest. Instead, each is employed in its optimal application scenario according to specific testing requirements, which reflects the current state of the sequencing field. The following provides a brief overview of several types of sequencing technologies, along with their advantages and disadvantages.
First-Generation Sequencing: The principle of Sanger sequencing involves using a DNA polymerase to extend primers annealed to the template sequence of interest until a chain-terminating nucleotide is incorporated. Each sequencing run consists of four separate reactions, each containing all four deoxynucleoside triphosphates (dNTPs) along with a limited amount of one specific dideoxynucleoside triphosphate (ddNTP). Since ddNTPs lack the 3'-OH group required for chain elongation, the growing oligonucleotides are selectively terminated at guanine (G), adenine (A), thymine (T), or cytosine (C) residues, depending on the corresponding ddNTP present in each reaction. The resulting fragments of varying sizes can be separated by high-resolution denaturing gel electrophoresis, thereby revealing the DNA sequence. Over years of development,First-generation sequencing is highly mature and currently serves as the international gold standard for all genetic testing.Clinically, nearly all DNA polymorphism validations are performed using Sanger sequencing.Second-generation and third-generation sequencing are also frequently validated using first-generation sequencing.Since 2010, although many next-generation sequencing instruments have emerged, first-generation sequencing continues to leverage its precision advantages for clinical testing and validation.
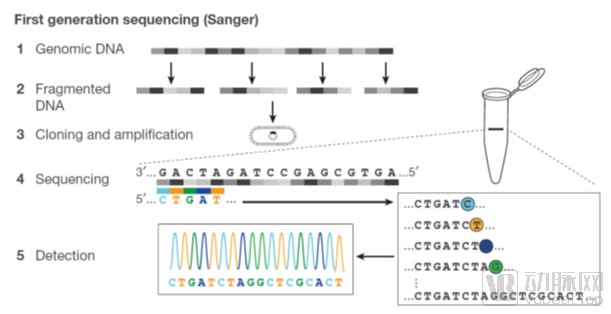
Sanger Dideoxy Chain Termination Method is a technique that generates four sets of nucleotide fragments of varying lengths, each terminating with A, T, C, or G, by initiating synthesis at a fixed point and randomly terminating at specific bases labeled with fluorescent dyes. These fragments are then separated by electrophoresis on urea-denatured PAGE gels to visualize and determine the DNA base sequence.
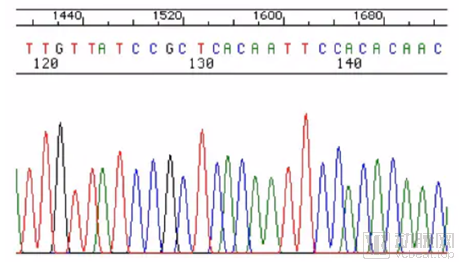
Figure of First-Generation Sequencing Results. The horizontal axis represents electrophoresis time and the sequential order of bases, while the vertical axis indicates fluorescence intensity. Higher and sharper peaks with minimal overlap between adjacent peaks signify greater accuracy in base calling. The results are visually apparent, intuitive, and accurate.
Next-Generation Sequencing: Second-generation DNA sequencing technology, also known as high-throughput sequencing (HTS), is characterized byLow Cost, High Accuracy, enabling rapid sequencing and analysis of hundreds of thousands to millions of DNA molecules from hundreds to thousands of samples simultaneously. Representative technologies of this era include Roche’s 454 (discontinued), Illumina’s Solexa (upgraded to NovaSeq, holding the largest market share), and ABI’s SOLiD (acquired by Thermo Fisher Scientific and upgraded to Ion Torrent S5). Due to the cutting-edge nature of sequencing technologies during this period, the market was predominantly monopolized by these three companies. The complexity of these sequencing technologies, the massive volume of generated sequencing data, and the high difficulty of subsequent bioinformatics processing have delayed their clinical adoption until only in the past five years, with applications primarily focused on precision oncology medication.
Taking Illumina as an example, we briefly introduce its sequencing workflow.
① Fragment the target DNA molecules into 100–200 bp segments, randomly ligate them onto a solid-phase substrate, and perform bridge PCR cycles involving Bst polymerase extension and formamide denaturation to generate abundant DNA clusters, each containing over 1,000 DNA fragments with identical sequences.
② The subsequent reaction is similar to the Sanger method, involving the addition of dNTPs labeled with four different fluorophores and incorporating reversible terminators. Each well on the solid-phase substrate contains eight independently detected sites, allowing for the parallel processing of eight independent libraries. It can accommodate millions of template clones, enabling the multiplexed detection of multiple samples, with each solid-phase substrate capable of reading up to 1 billion bases per run.
③ The DNA clusters hybridize with the universal sequences of single-stranded amplification products. Due to the action of terminators, DNA polymerase extends only one dNTP per cycle. The light signal generated by each extension is analyzed and sequenced by a standard microarray optical detection system. In the next cycle, the terminators and fluorescent labeling groups are cleaved off, allowing further dNTP extension, thereby achieving sequencing-by-synthesis technology.
④ Its main drawback is the short read length of sequences due to optical signal attenuation and phase shift, resulting in limited reading length for each DNA sequencing fragment. Currently, the most mainstream and cost-effective approach is paired-end 150 bp sequencing (PE150). Our strategy for whole-exome sequencing also employs PE150.
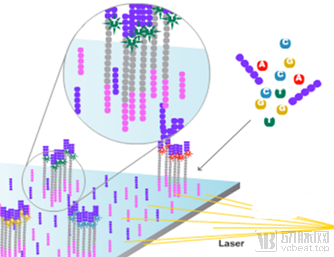
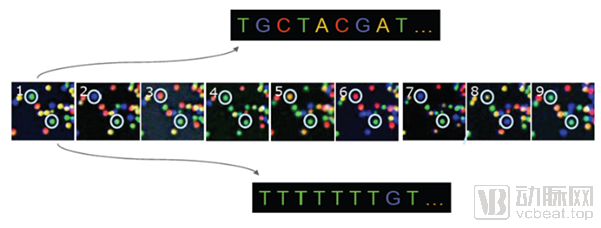
Principle of Next-Generation Sequencing: Determining DNA sequences by capturing, via CCD, the fluorescence emitted from dNTPs incorporated into templates during cluster generation.
Upon completion of next-generation sequencing (NGS), the resulting data are massive, complex, and heterogeneous, rendering the results invisible to the naked eye. Specialized algorithms and pipelines are therefore required to process raw data into usable information. This can be likened to the distinction between purchasing ingredients at a market (sequencing) and preparing them into a gourmet meal (algorithmic processing). This process demands high-performance computing resources, well-established robust algorithms, and personnel proficient in bioinformatics analysis.
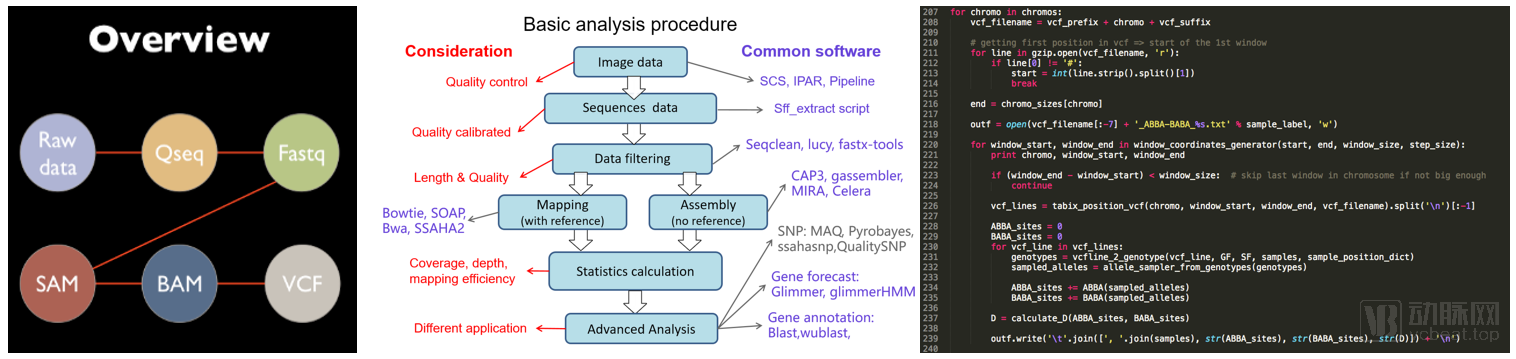 In the next-generation sequencing (NGS) analysis pipeline, raw sequencing data undergoes quality control to generate a series of intermediate files (left panel), which are then processed through complex bioinformatics workflows (middle panel) to ultimately assemble the target genes. Currently, the entire workflow is predominantly implemented on servers using Python and Perl (right panel).
In the next-generation sequencing (NGS) analysis pipeline, raw sequencing data undergoes quality control to generate a series of intermediate files (left panel), which are then processed through complex bioinformatics workflows (middle panel) to ultimately assemble the target genes. Currently, the entire workflow is predominantly implemented on servers using Python and Perl (right panel).
Due to inherent limitations in its underlying principles, next-generation sequencing (NGS) requires multiple reads of the same locus to ensure data reliability. For clinical applications, it is generally required that each locus be sequenced more than 200 times. This requirement is referred to as sequencing depth (>200×). NGS offers significant advantages, including massive data throughput, high-efficiency human genome sequencing within a short timeframe, and the ability to identify a large number of previously unknown loci. The reduced cost per locus lays the foundation for personalized medicine and precision pharmacotherapy.
Finally, there is another sequencing-like tool called gene chip. The prototype of gene chip was proposed in the mid-1980s. The sequencing principle of gene chips is based on hybridization sequencing methods. Currently, it is monopolized by several major manufacturers both domestically and internationally. Its advantages include a large amount of information, significantly increased recognition sites compared to first-generation sequencing, mass production capability, and fully standardized assembly line operations. However, its disadvantages are also obvious: the cost remains high, there is no technical barrier, it only detects pre-selected sites, and updating the sites involves significant time and financial costs. For example, Illumina's ASA chip integrates 660,000 human genome loci on a single chip, with an information detection capacity of approximately 30 million.
In 2001, the draft human genome was obtained through first-generation sequencing at a cost of $3.7 billion and over a period of 13 years. By 2007, completing the first complete map of the human genome sequence using second-generation sequencing cost only $1.5 million and took just three months. By 2020, human genome sequencing could be completed for less than RMB 10,000, requiring only three days. In recent years, sequencing technology has advanced by leaps and bounds. With the continuous decline in unit sequencing costs, we are bound to witness the day when everyone has their own “genetic ID card.”
3The Relationship Between Gene Mutations and Diseases
The vivid and figurative Chinese folk proverb, passed down for thousands of years, aptly describes the key characteristics of genes—Heredity and Variation。
“Dragons beget dragons, phoenixes beget phoenixes” reflects heredity; “the dragon has nine sons, each different from the others” results from genetic variation. Heredity and variation are ubiquitous and continuous phenomena in the biological world, serving as the foundation for speciation and biological evolution. The interplay of human genetic heredity and variation with environmental factors determines an individual’s birth, growth, aging, disease, and death. Modern medical research has demonstrated that nearly all diseases, except those caused by trauma, poisoning, or malnutrition, are associated with genes.
Diseases can be classified into at least four categories based on the extent to which genetic variations contribute to their pathogenesis: 1) Diseases determined entirely by genetics: such as congenital disorders found in monogenic rare genetic diseases, including osteogenesis imperfecta, hemophilia, and Duchenne muscular dystrophy. 2) Diseases primarily determined by genetics but requiring specific environmental triggers: for example, phenylketonuria (PKU), a monogenic genetic disorder. While early understanding linked it solely to heredity, it is now known that the disease is triggered only upon consumption of foods high in phenylalanine. 3) Diseases influenced by both genetic and environmental factors. This category is further subdivided into those predominantly determined by genetic factors, such as hereditary tumors and developmental intellectual disabilities; and those where genetic and environmental factors are equally significant, such as various chronic diseases including hypertension, coronary heart disease, and diabetes. 4) Diseases determined entirely by environmental factors: such as burns and scalds, and infections.
According to domestic and international guidelines and expert consensus, the relationship between diseases and genes can currently be simply categorized into five levels, including:
Pathogenesis:This generally refers to a causal relationship between gene mutations and disease pathogenesis, where results have been replicated across multiple populations and potentially validated through cellular and animal experiments.
For example, mutations in the ATP7B gene lead to impaired or absent ATPase function, resulting in reduced synthesis of serum ceruloplasmin (CP) and impaired biliary copper excretion. Copper ions accumulate and deposit in tissues such as the liver, brain, kidneys, and cornea, causing hepatolenticular degeneration (HLD). This condition was first described by Wilson in 1912 and is therefore also known as Wilson disease (WD). It is an autosomal recessive disorder of copper metabolism, characterized primarily by liver cirrhosis and basal ganglia damage due to copper metabolic dysfunction. Understanding of the pathogenesis of HLD has advanced to the molecular level. The global incidence of HLD ranges from 1/30,000 to 1/100,000, with approximately 1/90 individuals being carriers of the pathogenic gene. The disease is relatively common in China. HLD predominantly affects adolescents, with a slightly higher prevalence in males than in females. Without appropriate treatment, it can lead to disability or even death.
ContainsPathogenicityIndividuals with such mutations have a significantly higher risk of developing the disease compared to the general population. Homozygous pathogenic mutations are highly likely to directly cause the disease, while heterozygous pathogenic mutations have a 50% chance of being transmitted to offspring, resulting in different disease phenotypes depending on the mode of inheritance (dominant or recessive). Patients harboring these mutations should undergo specific testing for the associated conditions, and subsequent treatment plans should be formulated based on clinical findings.
Risk Factors:Generally, this refers to the significant presentation of gene mutations in the affected population, whereas such mutations are significantly reduced in the unaffected or non-onset population. Here, "significant" denotes a statistically significant difference in means. However, for such mutations, only statistical differences have been identified through population analyses stratified by different phenotypes; no etiological or pathological differences, nor the inevitable changes in signaling pathways, have been discovered. Furthermore, there is a lack of corresponding in vivo and in vitro experiments to validate their causal relationships.
For example, Microseminoprotein-beta (MSMB) is one of the three major proteins secreted by prostate epithelial cells. Eeles et al. (2008) conducted a genome-wide study on 1,854 patients diagnosed with prostate cancer before the age of 60 and 1,894 healthy controls with low PSA concentrations (less than 0.5 ng/mL), analyzing a total of 541,129 SNPs. They ultimately found that mutations at this locus in the MSMB gene were significantly associated with prostate cancer (8.7 x 10-29). The pathogenic T mutation affects the binding of numerous transcription-related factors. Thomas et al. (2008) conducted a genome-wide study involving 3,941 prostate cancer patients and 3,964 healthy controls, analyzing a total of 26,958 SNPs, and also found that the mutation at this locus in the MSMB gene was significantly associated with prostate cancer (7.31 x 10-13). Chang et al. (2009) conducted a study on 2,899 patients and 1,722 healthy individuals, finding that this locus in the MSMB gene had the strongest association with prostate cancer. Lou et al. (2009) investigated chromosome 10 in 6,118 European patients and 6,105 normal controls, also revealing that this locus in the MSMB gene showed the most significant association with prostate cancer (p = 8.8 x 10-18)。
Mutations classified as high-risk factors indicate a strong association between the specific genetic locus and the disease under investigation. The presence of such mutations constitutes a risk factor for disease onset and is correlated with a higher incidence rate. Patients harboring these mutations should prioritize relevant health management and medical care. However, due to limitations in current research depth, such mutations are not necessarily causative of the disease.
Uncertain:This indicates that findings regarding this genetic locus and the tested disease have been inconsistent across different studies, warranting further research to verify the correlation between alterations at this locus and disease pathogenesis;
Benign:Indicates that although this genetic locus is altered compared to the general population, the alteration is not associated with the disease under investigation;
Risk Unknown:This indicates that although this gene locus is altered compared to the general population, there are currently no studies reporting a correlation between alterations at this locus and the disease.
Through gene sequencing, we can clearly identify which variants in an individual’s genome are pathogenic mutations and which are risk-associated mutations, thereby enabling interpretation based on genetic testing results toAccurate Assessment of Disease Risk for Individuals and Their Offspring. Subsequently, personalized health management is implemented based on the assessment of these risks.
4Accuracy and Development of Disease Databases
However, these disease-associated mutations were not discovered out of thin air; it is only through the relentless, day-and-night research efforts of scientists that we have been able to uncover small glimpses into the previously unknown mechanisms underlying disease etiology.Therefore, we must establish a database that enables rapid retrieval of the aforementioned DNA variants associated with diseases, so as to complete the analysis of gene mutations and their disease associations within a short timeframe.
An accurate database can enhance the precision of analysis, facilitate the identification of disease etiology, and enable appropriate treatment. Conversely, an inaccurate database may lead to analytical errors, potentially resulting in subsequent misguided treatments. The following is an example of an inaccurate database:
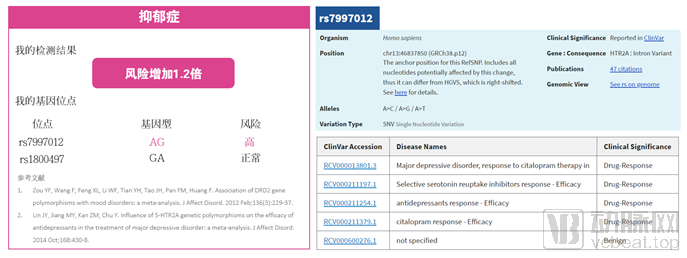
A report from a genetic testing company (left figure) indicates that the client is at risk for depression based on a heterozygous mutation at the SNP locus rs7997012. This conclusion is drawn from two cited articles, one published in 2012 and the other in 2014, both appearing in the Journal of Affective Disorders (impact factor: 3.5) and conducted as meta-analyses. Without detailing the algorithm used, we can first examine the genetic locus tested by the company: rs7997012. As shown in the right figure (sourced from NCBI), the clinical significance of this locus relates to the efficacy of antidepressant medications; mutations at this site alter drug response, rather than predisposing individuals to the onset of depression. Therefore, reliance on an incorrect database can lead to erroneous conclusions in genetic testing, even if the detected genotype is accurate.
If the client proceeds with subsequent depression treatment based on the aforementioned report, the outcomes may all be predicated on an erroneous judgment.So, how can one determine whether a database is reliable?
The current standard practice involves integrating several internationally recognized human genome and disease databases, such as the Online Mendelian Inheritance in Man (OMIM) database (www.omim.org/), the Human Gene Mutation Database (HGMD) (www.hgmd.cf.ac.uk/), and the clinical variant database ClinVar (www.ncbi.nlm.nih.gov/clinvar). Simultaneously, the latest authoritative scientific findings, particularly relevant articles published in top-tier journals such as Science, Cell, NEJM, JAMA, and Nature, are collected and curated. These integrated resources constitute a company’s core assets. Taking Zhunuo Cloud as an example, more than five dedicated personnel are required to build and maintain the disease database, including database architects and administrators, as well as scientists responsible for reviewing public databases and the latest authoritative literature. The workflow begins with screening reliable results from public databases; variants with high confidence ratings are incorporated into the company’s disease database. Scientists then conduct reviews to verify the biological and medical significance of these variants as described in the original literature. Variants that are problematic or lack statistical significance are excluded, while those with demonstrated significance are retained. Additionally, scientists performing these reviews collect and organize newly published literature, incorporating significant variants from these new studies into the company’s disease database. Finally, database architecture engineers organize these variant information points to create a comparable and queryable disease-related gene database.
This constitutes the core asset of every genetic testing company and is not easily subject to reasonable third-party valuation.
Finally, the databases of disease and gene mutations maintained by genetic testing companies or international public repositories are not static. Since the invention of PCR technology in the 1980s, our understanding of the relationship between gene mutations and diseases has gradually deepened. As discussed in Section 3, generally speaking, the more severe a disease is, the earlier its association with gene mutations tends to be identified, such as in β-thalassemia. In contrast, for milder chronic conditions, it is more difficult to identify associated genes; large-scale genome-wide association studies (GWAS) are required to gradually elucidate the relationship between genes and these diseases, such as hypertension and diabetes.
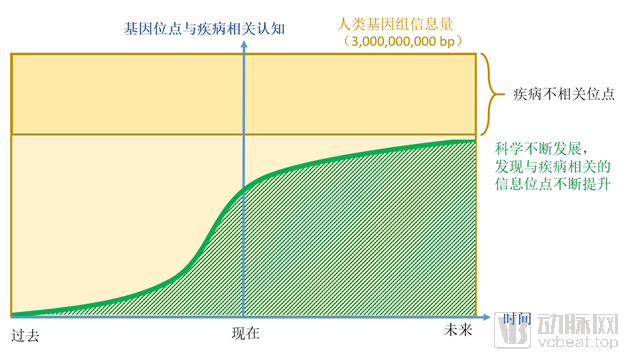
The correlation between human genetic loci and diseases is constantly evolving, and we are in a phase of continuously advancing understanding. Through past research, we have gradually elucidated the relationships between certain genetic mutations and diseases, primarily focusing on rare diseases and hereditary cancers. As time progresses and science continues to advance, an increasing number of genetic loci will be found to correlate with disease pathogenesis.
We are currently in a phase of continuously improving understanding of the correlation between genes and diseases. Therefore, although many gene mutations have been identified as potential causes of diseases, there is still a long way to go. With ongoing technological advancements, more disease-associated loci will be discovered, and the relationship between human genes and diseases will become increasingly clear.
5Summary
When revisiting the question, “Are genetic testing results accurate?”, the aforementioned discussion can be summarized as follows:
1)Are the sequencing results consistent with the actual DNA sequence?
Regarding the accuracy of sequencing,Under certain conditions,Whether it is the results from Sanger sequencing (with no overlapping peaks and a clean background), next-generation sequencing (ensuring sufficient sequencing depth and effective algorithmic processing), third-generation sequencing (ensuring sufficient sequencing depth and effective algorithmic processing), or microarray analysis (ensuring rational design and effective hybridization), all are accurate.The accuracy of genetic testing results can be reliably achieved across different platforms using various sequencing methods., but there are significant differences in the amount of valid information ultimately obtained by each technology.
2)Are the identified disease susceptibility loci truly associated with disease pathogenesis?
Generally speaking, if genetic testing reveals a risk of disease onset, the first step is to assess the pathogenicity level.Is it pathogenic? Or a risk factor? Or something else? When the aforementioned two types of loci are present, the current data are accurate.However, it is impossible for us to comprehend aspects that science has not yet reached. Therefore, from this perspective, the results should be viewed as follows:We cannot guarantee that the test is 100% accurate.. This includes: risk-associated loci identified may be refuted by future research; and loci identified as non-risk may later be found to confer risk as scientific research advances. Furthermore, the associations between a large number of yet-unknown genetic loci and diseases require further scientific development to provide more definitive evidence.
Therefore, under the premise that current databases are reliable, our detection of disease loci known to humansEnsure consistency with existing research, current genetic testing can ensure an accuracy rate approaching 100%. Nevertheless, it remains possible that existing authoritative findings could be overturned as larger population cohorts are incorporated into future studies. Therefore, genetic testing is a continuously evolving and lengthy process that requires greater participation from practitioners and the analysis of larger sample sizes. Only through such efforts can we establish a correct understanding of the relationship between diseases and genetic mutations in the future, thereby providing more robust answers to questions such as “Is genetic testing accurate?”