Germany Achieves Efficient Review: 15 Digital Therapeutics Approved Within 3 Months Under DiGA Fast-Track Program
From any perspective, digital therapeutics (DTx) have emerged as one of the recent innovations in the healthcare sector. However, this nascent field has also given rise to new challenges: an increasing number of companies are labeling themselves as “digital therapeutics” providers, yet Chinese regulatory authorities have not yet established specific approval requirements for DTx, leading to ongoing controversy regarding their classification and recognition.
Lessons from Abroad: Regulatory Approval of Digital Therapeutics in GermanyCountries and regions with earlier development and more advanced concepts in digital therapeutics have been implementing regulatory approval processes for these interventions for some time. Their regulatory authorities have accumulated substantial experience and lessons learned during this process and have issued corresponding guidelines, which offer valuable references. VCBeat (WeChat ID: Vcbeat) has compiled and summarized the regulatory approval landscape for digital therapeutics in Germany into this article.
This article will mainly introduce:Current Status of Digital Therapeutics Approval in Germany, How Germany Defines Digital Therapeutics, How Germany Distinguishes Between Health Apps and Digital Therapeutics, and the Fast-Track Approval Process for Digital Therapeutics in Germanyetc.
The Federal Institute for Drugs and Medical Devices (hereinafter referred to as BfArM, an abbreviation of the German Bundesinstitut für Arzneimittel und Medizinprodukte) is an independent higher-level authority under the German Federal Ministry of Health. Its role is similar to that of the U.S. Food and Drug Administration (FDA) and China’s National Medical Products Administration (NMPA). It is primarily responsible for drug licensing and safety improvements, testing and risk assessment of medical devices, monitoring of narcotic drugs, and regulating the legal trade in narcotic precursors.
The approval and certification of digital therapeutics clearly fall within the scope of BfArM’s responsibilities. In Germany’s medical device approval registry, a dedicated DiGA directory has been established specifically for digital therapeutics. DiGA is an abbreviation of the German term “Digitale Gesundheitsanwendungen,” meaning Digital Health Applications (hereinafter, DiGA will be used to refer to German digital therapeutics). The BfArM recognizes that DiGAs encompass a broad range of possibilities, such as disease diagnosis and treatment, as well as self-directed healthy lifestyle habits, holding promise to become digital assistants at patients’ fingertips.
VCBeat interviewed Dr. Chen Suyuan, Founder of Maibo HealthTech, and Ms. Zuo Simin, Co-Founder, regarding certain details of the German DiGA approval process. Since its inception, Maibo HealthTech has been actively promoting Sino-European scientific and technological exchanges and cross-border project collaborations. Focusing on the frontiers of healthcare and clinical needs, the company provides clients with industry insights, a platform for cross-border corporate exchanges, and business and technical consulting services.
Dr. Chen Suyuan believes that the rise of Germany’s Digital Health Applications (DiGA) is largely driven by two factors. “On one hand, Germany faces long-term challenges from an aging population, leading to growing demand for chronic disease management; on the other hand, digital health technologies are rapidly advancing, yet there is a lack of specialized laws and regulations for oversight.”
As DiGA cannot be entirely equated with traditional software application approvals, after a period of exploration and under the guidance of the spirit of Section 139e of the German Social Code Book V: Statutory Health Insurance (Fünftes Buch Sozialgesetzbuch, abbreviated as SGB V), the BfArM has designed a dedicated fast-track approval process for DiGA (The Fast-Track Process for DiGA).
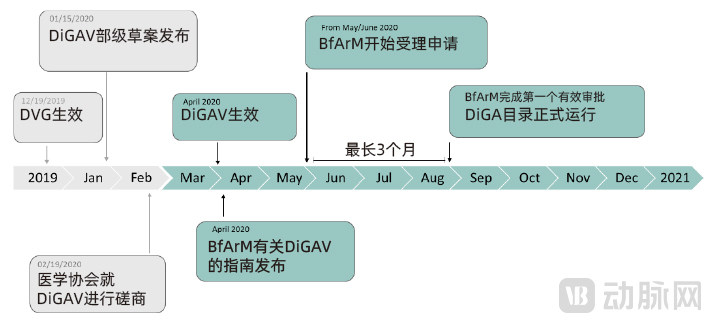
On May 27, 2020, the DiGA fast-track approval process was officially implemented, and just over a year has passed since then. As of June 2, 2021, a total of 75 applications had been submitted through the DiGA fast-track approval process. Among these, 52 applications were for provisional listing, while 23 were for permanent listing. Of the 75 applications, 15 were approved, 4 received negative evaluations, 33 were voluntarily withdrawn by their developers after submission, and 23 remained under review.
Dr. Chen Suyuan believes that the fast-track approval process for DiGA has had multifaceted impacts. Germany’s universal health insurance system possesses strong payment capacity, and the passage of the Digital Healthcare Act (DVG) has provided robust momentum for the development of the DiGA market, a move welcomed by relevant companies. Although health insurers are required to cover these costs, they also welcome the initiative due to considerations of promoting market standardization and fair competition.
Relatively speaking, both physicians and patients maintain a cautious stance. “Many physicians are unfamiliar with this approach and hold a rather conservative view toward DiGA (Digital Health Applications). They remain uncertain about the extent to which DiGA can benefit patients. Furthermore, they are concerned that regulatory frameworks may be incomplete, potentially leading to some degree of market disorder as more companies enter the space.”
“As for patients, although they are excited about the potential of DiGA to aid in disease management and treatment, they also wish to understand what outcomes a DiGA can achieve and to what extent it can improve health,” added Ms. Zuo Simin.
Simply put,The BfArM considers DiGAs to be Class I and Class IIa medical devices certified with the CE mark in accordance with the European Union’s Medical Device Regulation (hereinafter referred to as “MDR”) and the Medical Device Directive (hereinafter referred to as “MDD”), possessing the following characteristics: the medical function of a DiGA shall be primarily delivered through its digital software capabilities; it may support the identification, monitoring, treatment, or alleviation of diseases, or the identification, monitoring, treatment, alleviation, and compensation of physical injuries or disabilities; and it may be directed solely at patients or simultaneously at both patients and healthcare institutions.
It is evident from the definitions that CE marking and DiGA approval are distinct. Simply put, obtaining CE marking does not guarantee approval under Germany’s DiGA framework; however, any digital health application (DiGA) approved by the BfArM must have already obtained CE marking.
“At present, there is no unified regulatory framework for digital therapeutics at the EU level, but I believe this is the direction of future development. From the perspective of medical device or drug approval in the EU, it is actually based on the entire EU, and after passing the EU’s approval, it enters the member states. Digital therapeutics are relatively new, and currently only Germany has a specialized approval process for them. I think that in the future, Europe will definitely introduce universal regulations, allowing entry into the entire EU market through a unified application procedure.” Dr. Chen expressed optimism about the prospect of unified legislation at the European level.
Timeline of Germany's DiGA Approval Policy
It is worth noting that the BfArM established the fast-track approval process for DiGAs during the transition period between the Medical Devices Directive (MDD) and the Medical Devices Regulation (MDR). The MDR officially came into effect on May 26, 2021. Subsequent changes to the definition or classification of medical devices may occur to comply with the new MDR requirements.
DiGA: More Than Just a Software Application, It Is a Medical Device
From the definition of DiGA, medical devices are the primary consideration. To facilitate the rapid distinction between general health and wellness applications and software applications classified as medical devices for both the public and developers, the BfArM has established detailed and practical guidelines. Based on whether a software application is definitively classified as a medical device, two scenarios can be distinguished. Furthermore, once a software application is recognized as a medical device, it must meet the same fundamental regulatory requirements in its manufacturing and production processes as traditional integrated research-and-production medical devices, without exception.
The first category comprises applications that can be definitively classified as medical devices. Under the German Medical Devices Act (hereinafter referred to as “MPG,” the abbreviation of the German term *Medizinproduktegesetz*), software applications such as smartphone apps may be classified as medical devices provided they are intended for use in humans and serve at least one of four specified purposes.
These four categories are: software applications used for the diagnosis, prevention, monitoring, treatment, or alleviation of disease; used for the diagnosis, monitoring, treatment, alleviation, or compensation of physical and mental injuries or physiological defects; used for the investigation, replacement, or modification of anatomical or physiological processes; and used for controlling conception.
Overall, the BfArM considers that any standalone software involving intervention based on data or information can be classified as a medical device of a certain class. However, this does not include software used solely for providing knowledge or information; for example, electronic instructions for use are clearly not medical devices.
The second category comprises applications that may be classified as medical devices. The intended uses claimed for these applications generally include the following terms: alarm, analysis, calculation, detection, diagnosis, interpretation, conversion, measurement, control, monitoring, and amplification.
Among these, software applications that can be classified as medical devices of a certain level mainly include the following categories: clinical decision support applications represented by those providing therapeutic measures; applications for calculating drug injection dosages (excluding simple methods that merely replicate a table to facilitate user inference of injection dosage); and applications that monitor patients and collect data, where the monitoring results have a significant impact on diagnosis or treatment.
It should be emphasized that mere data storage, archiving, lossless compression, communication, or simple information search applications cannot be classified as medical devices. For example, software specifically designed for data archiving is clearly not a medical device.
Based on a simple classification by software intended use, decision support software, software systems, telemedicine systems, Hospital Information Systems (HIS), and Picture Archiving and Communication Systems (PACS) are classified as medical devices. Operating system software and general-purpose software are not considered medical devices. As for whether health software applications are classified as medical devices, this depends on determining if they are intended for medical purposes; if they are used solely for exercise, fitness, or nutritional goals, and the developer claims no medical purpose, they are generally not considered medical devices.
Interestingly, some developers may attempt to circumvent regulations by employing “gray-area” tactics, such as simply stating “This is not a medical device” in their app store descriptions. However, under the German Medical Devices Act (MPG), if a software application states or implies an intended medical purpose in any public materials—including labels and instructions for use—it will still be classified and regulated as a medical device by the Federal Institute for Drugs and Medical Devices (BfArM).
As a software application, DiGA must strictly comply with relevant data security regulations. The European Union is one of the regions with the most stringent data security requirements globally and where citizens place the highest value on personal privacy. Therefore, DiGA must adhere rigorously to applicable data security laws and regulations. If DiGA involves data processing outside Germany, it must comply with the corresponding legal and regulatory frameworks. Even DiGA products that have received formal approval may require technical adjustments in response to updates in data and privacy security standards. Failure to do so may result in the revocation of existing approvals in accordance with the law.
“Due to the COVID-19 pandemic, an increasing number of people have begun to adopt relevant DiGA applications. This inevitably raises a critical issue: data security and privacy protection. I believe this is not only a concern for DiGA but also a mandatory consideration for any future related products or services seeking to enter the German market,” emphasized Ms. Zuo Simin, highlighting that data security and privacy protection are accorded high priority in Germany.
DiGAs must not pose any potential risk of serious harm to the human body.
The reason DiGA includes only software applications classified as Class I and Class IIa medical devices is based on risk considerations. In the European Union, except for in vitro diagnostic medical devices and active implantable medical devices, medical devices are categorized into different risk classes based on the potential harm that may result from their errors or malfunctions, ranging from Class I (low risk) to Class IIa or IIb (medium risk), and up to Class III (high risk).
Under the provisions of the Medical Device Directive (MDD), among the several rules applicable to determining whether a software application qualifies as a medical device (primarily including Rule 9, Rule 10, Rule 12, Rule 14, and Implementation Rule 2.3), medical devices classified as Class IIb may have potential and irreversible significant impacts on human health in certain ways.
For example, Rule 10 stipulates that active devices used for diagnosis and meeting the criteria of “providing energy to be absorbed by the human body (excluding equipment used to illuminate the patient’s body in the visible spectrum), imaging the in vivo distribution of radiopharmaceuticals, or directly diagnosing or monitoring important physiological processes of the human body” are classified as Class IIa medical devices.
However, once the physiological signs monitored by such active devices that directly diagnose or monitor critical human physiological processes are of vital importance and may directly pose a life-threatening risk to patients (such as electrocardiographic parameters, respiration, and central nervous system activity), they shall be classified as Class IIb medical devices. In addition, interventional radioactive active devices that generate ionizing radiation for diagnostic and therapeutic purposes, as well as devices (including software applications) used to control or monitor such equipment or that directly affect their performance, are also classified as Class IIb.
Under the MDD rules, medical applications on smartphones and tablets will be classified as Class I medical devices. However, if they are used for diagnosing or monitoring vital signs (such as ECG parameters), they may be classified as Class IIa or IIb medical devices depending on the level of risk. Once classified as a Class IIb medical device, the application will not be recognized as a DiGA and will be subject to more stringent regulatory requirements.
In other words, the BfArM explicitly holds that DiGA must demonstrate a high level of safety and must not pose potential risks to human health. Software applications that, in the event of malfunction, could place users in potentially dangerous situations cannot be classified as DiGA.
Furthermore, software that merely serves to collect and transmit device data or to control devices cannot be classified as a DiGA; applications used exclusively by physicians for patient treatment are also not considered DiGAs. A DiGA must involve interactive engagement with the patient; therefore, solutions that solely collect data from sensors or mobile devices and transmit it directly to physicians do not meet this requirement.
Sometimes, DiGA does not appear as a standalone software application but is combined with other hardware, software, or services. In some cases, such combinations can be classified as DiGA, and regulatory approval may even be mandatory; in other cases, this is not the case. Based on past experience, the BfArM has provided a detailed analysis of the respective possibilities.
Integration with Other Software, Hardware, and Services
DiGAs come in various forms, ranging from common mobile apps to desktop or browser-based applications on computers. They may also be combined with hardware or software, provided that their primary function is delivered through the digital technology of the software application. The BfArM has provided very specific guidance on whether the combined use of DiGAs with hardware and software qualifies as a DiGA.
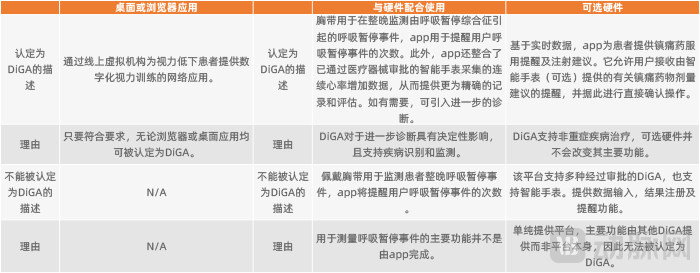
Case Studies on the Classification of DiGA Integrated with Software and Hardware
DiGA can also be combined with additional services. In principle, additional services such as consulting, training, or those provided by commercial insurance companies may be offered by DiGA or integrated into its use. It must be clarified that such additional services will not be reimbursed by statutory health insurance. Given that DiGA is eligible for reimbursement by statutory health insurance, the documentation demonstrating the positive medical effects of the software application, which is required for approval, must not include these additional services. Furthermore, developers need to list all application scenarios involving additional services in their communications with the BfArM to avoid any misunderstandings.
Of course, the aforementioned situation does not apply to services provided by physicians certified under the statutory health insurance system. Services related to the intended use of DiGA, when delivered by health insurance-certified providers—including attending physicians, resident physicians, dentists, or psychiatrists—are reimbursed by the statutory health insurance as physician services. Therefore, these services can be included as part of the documentation required for DiGA approval.
If the services provided by physicians accredited under the statutory health insurance scheme can be described in text as part of the DiGA usage process, or if the EBM codes required for reimbursement can be listed directly (in most cases, each reimbursable service under the German statutory health insurance system has its own independent EBM code). It should be noted that these services must be provided by physicians accredited under the statutory health insurance scheme and supported by submitted documentation for reimbursement; otherwise, they will not be eligible for coverage by the statutory health insurance.

Case Studies on the Certification of DiGA Integrated with Services
Regarding the relationship between internet-based healthcare and DiGA, the BfArM holds that if the primary functions are delivered through software-based digital technologies, then the internet-based healthcare application constitutes part of a DiGA; however, a pure internet-based healthcare platform cannot be classified as a DiGA. In terms of scope, DiGA encompasses more than internet-based healthcare (i.e., DiGA > internet-based healthcare). Of course, individual exceptional cases must be determined in consultation with the BfArM.
Scope Determination of DiGA When Integrated with Other Functionalities
In some cases, DiGAs may offer additional optional services or features, such as integration with social media platforms, compatibility with supplementary hardware and software, or inclusion of modules that have been separately approved as medical devices. In such scenarios, how should the regulatory boundaries of DiGA approval be delineated?
The BfArM holds that the boundary lies in ensuring that these additional features or services do not exert any influence on the intended medical functions submitted during the DiGA approval process, nor do they compromise or alter their positive medical impact. Furthermore, these features should remain relatively independent of the DiGA, such that any malfunction in these features does not affect the operation of the DiGA itself.
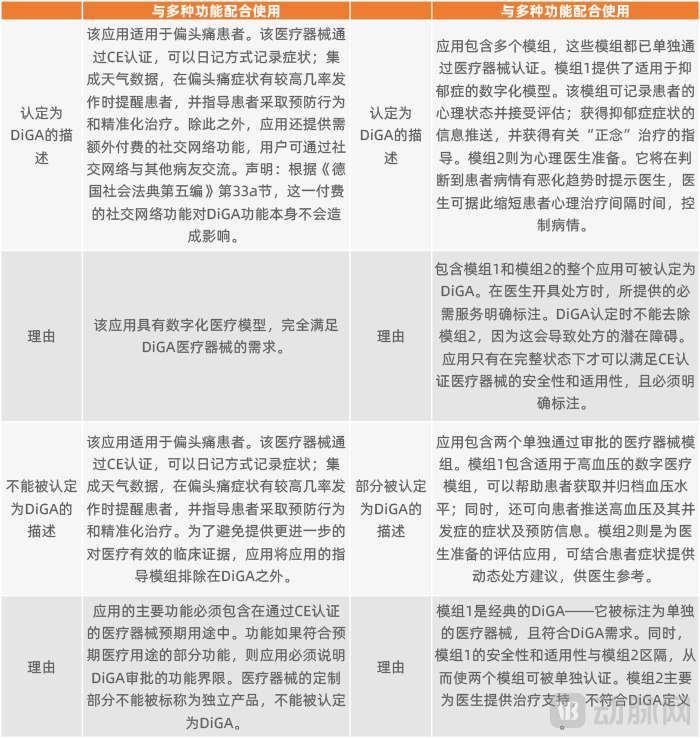
Examples of DiGA Recognition Combined with Additional Features
In accordance with relevant regulations, the BfArM currently does not approve additional features. Naturally, if these additional features require payment, the costs must be borne entirely by the users out-of-pocket and are not covered by statutory health insurance. Therefore, such additional features must be clearly labeled separately, with an explicit statement that they are not part of the DiGA-certified functionalities.
Can software applications used for disease prevention be classified as DiGA?
Whether software applications used for disease prevention are classified as DiGA depends on the specific circumstances. Within the German healthcare system, disease prevention is categorized into several levels. Primary prevention, the most fundamental level, targets the general population to prevent the onset of diseases. In short, primary prevention applies to healthy individuals. It promotes healthy lifestyles through specific formats (such as health courses) and serves as a tool for health assessment.
However, under the current definition, DiGAs are used to “support the recognition, monitoring, treatment, or alleviation of diseases, or the recognition, monitoring, treatment, alleviation, and compensation of physical injuries or disabilities.” It is explicitly stipulated that they do not include functions for disease avoidance or prevention and are not applicable to healthy populations. Therefore, software applications intended solely for primary prevention clearly do not fall within the scope of DiGAs.

Certification Case of DiGA Integrated with Smart Scales
Software applications designed for secondary prevention, which primarily aims to prevent disease progression, and for tertiary prevention, which aims to prevent complications and comorbidities, meet the definition of “disease treatment” under DiGA. However, approval is contingent upon the disease having specific risk factors that can be coded into specific disease codes using a disease classification system.
Germany believes that digital therapeutics, as an emerging category of medical devices, will systematically unlock the innovative potential of digital health applications in the country. On December 19, 2019, Germany’s Digital Healthcare Act (hereinafter referred to as the DVG, or Digitale-Versorgung-Gesetz) came into effect. This legislation laid the legal foundation for the subsequent approval of digital therapeutics in Germany, marking the integration of patient-prescribed “apps on prescription” into the national healthcare system.
To better align with the inherent characteristics of Digital Health Applications (DiGA), the German Federal Ministry of Health has established provisions regarding the application process, DiGA requirements, and the DiGA Directory in the supplementary legal regulation known as the “Digital Health Applications Ordinance” (abbreviated as DiGAV, or Digitale-Gesundheitsanwendungen-Verordnung). DiGA is regarded as an integral component of digital healthcare. Consequently, the compliance requirements set forth by the DiGAV necessitate that DiGA developers integrate their solutions in parallel with Germany’s evolving nationwide electronic health infrastructure.
The BfArM provides corresponding guidelines for DiGA approval in accordance with the provisions of Book V of the German Social Code (Statutory Health Insurance). The BfArM has specifically designed a fast-track approval process for DiGAs, with the details of this procedure regulated by the German Federal Ministry of Health. Once a software application is approved by the BfArM, becomes a recognized DiGA, and is included in the DiGA Directory, it may be prescribed by physicians authorized under statutory health insurance for patients with indicated conditions, with reimbursement covered by statutory health insurance.
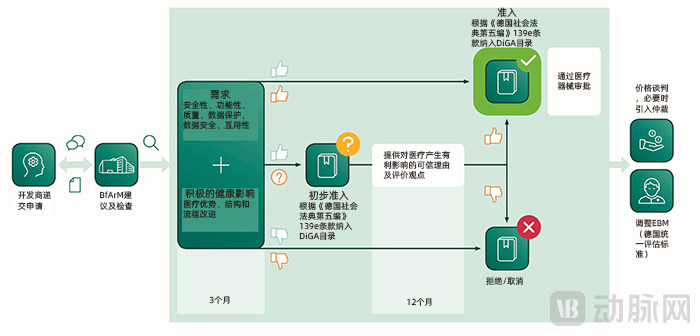
General Application Process for DiGA
From the moment developers submit an application for a software application, the BfArM must evaluate the applied-for DiGA within three months. By reviewing statements regarding product quality (such as data protection, interoperability, and user experience) and assessing evidence of the medical benefit of the corresponding software application (i.e., its impact on improving users’ health status or managing their conditions), the BfArM will determine whether the DiGA application is approved.
Information Included in the DiGA Directory
Approved DiGAs will be listed in a dedicated DiGA Directory, ensuring that all stakeholders within the German healthcare system can access information relevant to their needs. This body of information includes not only details on the medical benefits of the listed DiGAs and related statutory health insurance reimbursement information, but also data on compliance with software data protection requirements and medical device regulations. These information items are categorized into five groups in total.
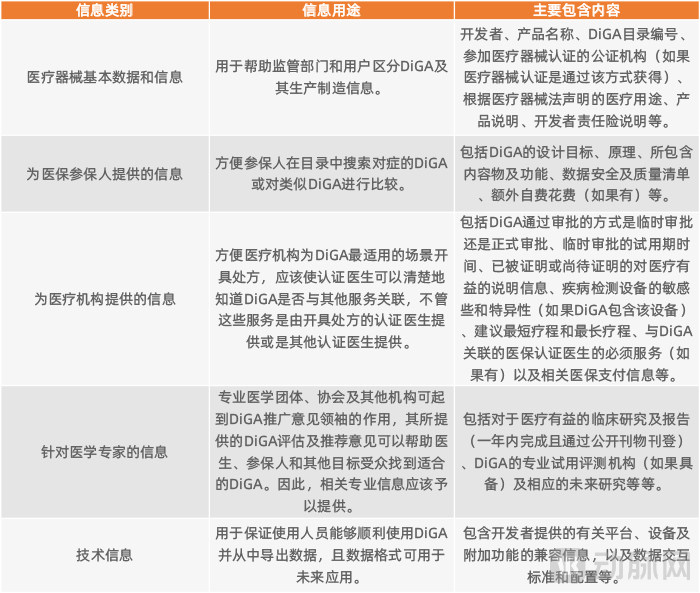
Key Information Included in the DiGA Directory
Difference Between Formal Approval and Provisional Approval for DiGA
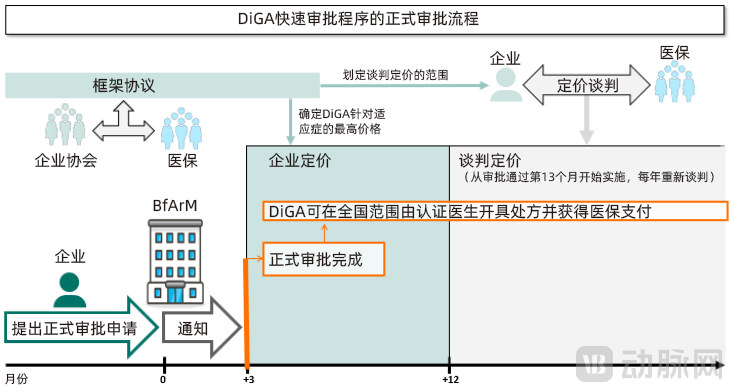
When applying for DiGA status, developers must decide whether to seek full approval or provisional approval. This decision largely depends on whether the applicant can provide comparable clinical data demonstrating the medical benefit of the DiGA.
If comparative clinical trials demonstrating the medical benefits of a DiGA have been completed at the time of application, formal approval may be applied for. If the clinical trial data are accepted, the BfArM will complete the approval process and include the DiGA in the directory within three months from the date of application.
If the initial application for formal approval fails due to certain reasons (e.g., clinical studies demonstrating the medical benefits of the application are not recognized), the applicant must reapply no earlier than 12 months from the date of rejection and must submit new data. The applicant may not immediately apply for provisional approval following the rejection of the formal application.
Of course, if applicants withdraw their applications on their own initiative before the BfArM makes a final decision, they can avoid this outcome. Consequently, in the approval status disclosed by the BfArM, nearly half of the applicants voluntarily withdrew their applications. The BfArM recommends that applicants who are uncertain whether their upcoming submissions for medically beneficial research will meet the requirements may seek prior consultation.
If the applicant has not yet completed comparable clinical trials, they may apply for provisional approval. During the probationary period, the applicant must complete comparable clinical trials and submit corresponding evidence demonstrating the medical benefits of the DiGA. The duration of the probationary period is determined by the BfArM and shall not exceed 12 months. Within three months of receiving these clinical data, the BfArM will decide whether to convert the current provisional approval into full approval.
If a comparable clinical trial report demonstrating the medical benefit of the DiGA is not submitted within the specified timeframe, or if the report fails to pass review, the BfArM will revoke the provisional approval of the software application and remove it from the directory. Once removed from the directory, the developer may only submit a new application 12 months after the date of removal, and must provide clinical data that differs from the previously submitted data. Applications based on repeated clinical data submissions will be rejected.
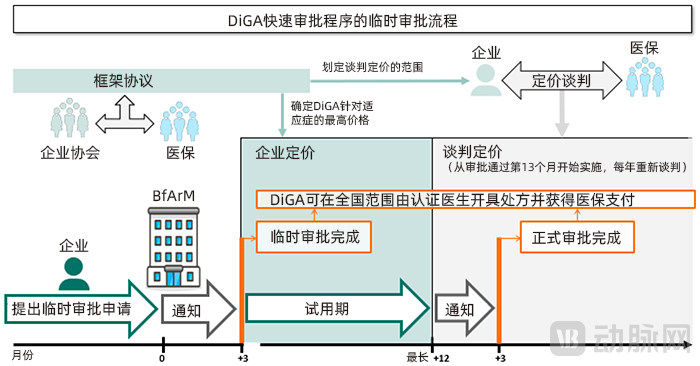
Generally, reimbursement under provisional approval is lower than that under formal approval. When applying for provisional approval, applicants must negotiate with the statutory health insurance funds to determine the maximum reimbursement amount for the first year following formal approval; meanwhile, applicants are also required to propose a lower reimbursement cap for the trial period. Additionally, developers can receive reimbursement provided by law during the DiGA provisional approval trial period, but the costs associated with concurrent clinical studies must be borne by the developers themselves.
It is worth noting that the BfArM emphasizes that it only reviews the software application itself as submitted for approval; once approved, it becomes eligible for reimbursement under statutory health insurance. Whether an approved DiGA also has versions with different business models (such as the common ad-supported free model) does not affect the approval process.
In exceptional cases, provisional approval allows for a single application to extend the trial period. The duration of the extension is determined by the BfArM and may not exceed 12 months. An extension is only granted if the clinical data for the DiGA has been submitted in accordance with regulations, and the BfArM determines based on this data that there is a high probability of medical benefit, even though the final assessment and report are not yet complete. Applicants seeking an extension must submit their request at least three months before the original trial period expires to allow sufficient time for review.
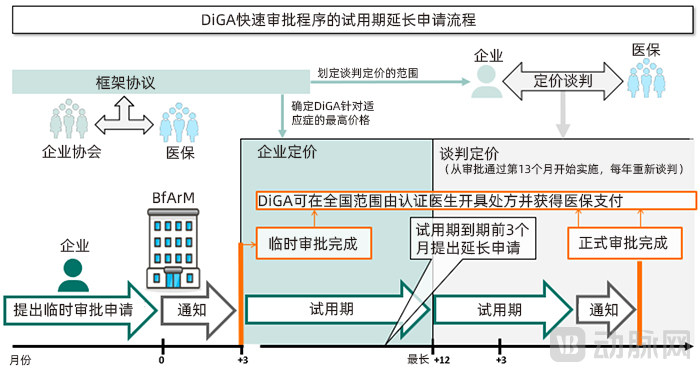
The BfArM guidelines explicitly list the documentation required for DiGA approval, including a checklist of software applications that meet DiGA approval criteria, descriptions of the safety and suitability of the software applications, data protection, information security, interoperability, and additional quality requirements.
Meeting these requirements is not difficult for the vast majority of applicants. Therefore, the key determinant of whether a software application can secure approval and be designated as a DiGA lies in the persuasiveness of the evidence submitted to demonstrate its positive impact on healthcare.
How is “evidence of the medical benefit of DiGA” defined?
"Social Code Book V: Statutory Health Insurance" of Germany provides a corresponding definition for "evidence of medical benefit of DiGA." The DVG and DiGAV further clarify the definition: evidence of medical benefit can be reflected in either medical benefits or improvements in the structure and processes of patient-related healthcare services. Software applications need to demonstrate that the DiGA has at least one piece of evidence of medical benefit.
The medical benefits are primarily reflected in improvements in users’ health status, shortened disease duration, extended life expectancy, and enhanced quality of life. The materials must be presented from the patient’s perspective, with particular emphasis on reductions in morbidity and mortality rates, or improvements in quality of life.
Structural and procedural improvements in patient-related medical services form the basis of reimbursement under the German health insurance system. These improvements primarily encompass the following components: diagnosis, monitoring, treatment, or alleviation as part of disease management; diagnosis, treatment, alleviation, or compensation for physical and psychological injuries or physiological impairments; support for patients in adopting healthy lifestyle habits, or integration of healthcare institutions and patients into streamlined care processes; coordination of treatment pathways, including specific implementation guidelines and standards to ensure therapeutic efficacy; reduction of barriers to accessing relevant medical services; and mitigation of daily life inconveniences caused by illness for patients, as well as reduction of the caregiving burden on patients and their families.
In principle, DiGA should be able to enhance its status in healthcare by providing information, enabling direct participation, or assisting in decision-making, and reduce the consumption of resources for treatment through the means provided by DiGA.
In the DiGA approval process, evidence for the aforementioned two aspects is mandatory. Additionally, applicants may submit information on potential healthcare benefits spanning both domains, which could positively influence future statutory health insurance reimbursement under specific conditions. This submission is optional and at the applicant’s discretion; however, if provided, the relevant documentation must still comply with applicable regulations.
Use ICD-10 to define the patient population eligible for DiGA
The BfArM requires that DiGA applications clearly define the applicable patient population, and data demonstrating medical benefit must be derived from at least one of these defined populations. Certified physicians may only prescribe DiGAs to these patient groups and receive reimbursement from statutory health insurance funds accordingly.
The BfArM requires the use of ICD-10 codes to define and differentiate the applicable patient populations. Consequently, once approved, a Digital Health Application (DiGA) can seamlessly integrate with various systems that utilize ICD coding—such as Diagnosis-Related Groups (DRG) and outpatient reimbursement schemes—and precisely distinguish the target patient groups. For instance, this approach makes it immediately clear whether a specific DiGA is intended for all patients with type 2 diabetes or only for those with type 2 diabetes accompanied by specific complications.
From this, we can also see that Germany’s emphasis on information technology systems in healthcare indeed covers all aspects and demonstrates strong practical applicability, making it highly worthy of reference for China.
Comparable Clinical Evidence
To demonstrate the medical benefits of the software application under application, developers must provide results from controlled experiments proving that using the applied-for software yields better outcomes than not using it. The experimental group should incorporate the applied-for software as part of the treatment for the patient population, while the control group should employ various control methods (such as no treatment, treatment without the applied-for software, or treatment using other approved DiGA) for comparison.
Depending on specific requirements, the study may be either a clinical study or an epidemiological study. Furthermore, research methods originally designed for other scientific fields (such as medical research, social research, or behavioral research) are also acceptable, provided they are appropriate for the selected field and constitute quantitative comparative studies. In this context, the use of real-world study data is permitted.
These studies must meet the requirements for DiGA approval in multiple aspects. First, the studies conducted must be implemented within Germany. Of course, in individual cases, if comparative evidence applicable to the medical scenario can be provided, data obtained outside of Germany may also be recognized.
Second, comparative studies must be registered with a compliant public registry. Compliance means that the registry must be a primary or partner registry of the WHO International Clinical Trials Registry Platform (ICTRP), or a data provider to this platform. In Germany, this typically refers to the German Clinical Trials Register (DRKS), which operates under the Federal Institute for Drugs and Medical Devices (BfArM). Completed clinical trial data may also be submitted to this register.
Third, the research should comply with relevant internationally recognized standards to facilitate its future use for demonstration and research purposes. Applicants are required to submit at least one retrospective comparative study, such as a case-control study, a retrospective cohort study, or a within-subject comparative study. At any time, applicants may also submit prospective comparative studies, which inherently provide a higher level of evidence.
Third, research data must be publicly published and comply with the publication standards for relevant academic papers. Corresponding data submissions must also be made in accordance with the requirements of the BfArM. For instance, the BfArM mandates that negative data from clinical studies be made public; furthermore, data from corresponding clinical studies must be submitted to the BfArM before they are publicly released and prior to their acceptance by a clinical study registry.
Required Documents for Temporary Approval
Compared to the formal approval process, the provisional approval pathway does not require the submission of clinical evidence demonstrating positive healthcare effects. However, applicants must still provide a plausible justification that the Digital Health Application (DiGA) can achieve at least one potential positive healthcare effect for specific patient populations. Applicants may submit systematic literature reviews and evaluations, or systematic evaluation data generated by the software application itself. The Federal Institute for Drugs and Medical Devices (BfArM) will use this information as the basis for determining whether it supports granting provisional approval.
The applicant must also submit an evaluation protocol developed in accordance with generally accepted scientific standards, which appropriately takes into account the results of data assessments. Among these documents, at least one must be provided by an independent third-party research institution, explaining how the proposed design yields the evidence of medical benefit required for formal approval. The independent third-party research institution must have no conflict of interest with the applicant, although it may charge corresponding fees based on market standards.
As previously mentioned, DiGAs granted provisional approval may submit a single application to extend the trial period by up to 12 months to meet the requirements of clinical studies. This is permissible only if the BfArM, based on existing data, determines that it is highly likely the study will yield conclusions beneficial to patient care.
When applying for an extension of the probationary period, the applicant must also provide justified reasons explaining why evidence of medical benefit could not be provided during the Phase I trials. Meanwhile, if required, the applicant may also need to provide justified reasons explaining why the extended probationary period can generate the actually missing evidence.
In the most favorable scenario, a DiGA granted provisional approval may be eligible for a trial period of up to 24 months. However, since many DiGAs exert their effects by subtly modifying users’ lifestyle habits, some stakeholders still believe that a two-year window may be insufficient to demonstrate their medical benefits. In response, the BfArM notes in its guidelines that, in principle, studies can be initiated prior to the DiGA application, thereby effectively addressing this concern.
Currently, Germany has taken the lead in providing comprehensive approval guidelines for digital therapeutics, along with a dedicated fast-track approval pathway. Dr. Chen Suyuan and Ms. Zuo Simin believe that, compared to the approval processes for digital therapeutics in other countries or regions, Germany’s DiGA fast-track procedure is characterized by its comprehensiveness, speed, and professionalism.
First is comprehensiveness. Although most countries or regions have some regulations related to medical health applications, none can provide such a detailed approval process that spans from legislation to rules and even covers various exceptions. In this regard, the comprehensiveness and completeness of Germany's DiGA approval process are second to none.
Second is the streamlined approval process. Germany’s DiGA approval framework fully accounts for the rapid update cycles inherent to software applications, allowing approval to be completed within three months of application submission (provided all documentation is complete). This represents a significant advantage for numerous small and medium-sized enterprises (SMEs), substantially reducing both time and financial costs.
Next is professionalism. The German DiGA approval process places particular emphasis on the medical attributes of digital therapeutics, regarding evidence of clinical benefit as the most critical supporting material. It imposes relatively stringent requirements ranging from study design to final submission, thereby ensuring that approved DiGAs are indeed beneficial to patients’ health.
“We have compared Spain and the UK, neither of which has established a separate approval pathway for medical apps distinct from medical devices; even the United States lacks a corresponding specialized process. I believe this is a highly innovative move by Germany.” Dr. Chen Suyuan considers this one of the most distinctive features of Germany’s DiGA approval framework.
Finally, payment is convenient. Once approved, DiGAs can quickly enter the market, depending on the completeness of their preparatory support work (even providing ICD codes for indications), where certified physicians can prescribe them to patients with the indicated conditions, and the costs are covered by statutory health insurance.
Digital therapeutics have been accompanied by both attention and controversy since their inception, with the root of the debate lying in whether they are beneficial to healthcare. The resulting public skepticism has given rise to the practical question of who should bear the cost. Germany’s approval framework for digital therapeutics represents one approach to addressing this issue: by defining them as a special class of medical devices, subjecting them to targeted and rigorous review, and incorporating them into statutory health insurance coverage post-approval, thereby alleviating public concerns. This approach may offer valuable lessons for China.