Navigating the EU MDR: Enhanced CE Certification Value and Strategic Pathways for Chinese Medical Device Exporters
On May 5, 2017, the European Union (EU) issued the new Medical Device Regulation (MDR) (EU 2017/745) to replace the old Medical Devices Directive (MDD) (93/42/EEC) and the Active Implantable Medical Devices Directive (AIMDD) (90/385/EEC). It came into effect on May 26 of the same year, with a three-year transition period. During the transition period, medical device manufacturers could voluntarily choose to apply for CE certification under either the old MDD or the new MDR; upon expiration of the transition period, compliance with the new MDR became mandatory.

Timeline of the EU Medical Device Regulation (Graphic by VCBeat)
However, the new Medical Device Regulation (MDR), originally scheduled for mandatory enforcement on May 26, 2020, was postponed by one year due to the impact of the pandemic and other related factors, and only came into full force on May 26 this year.
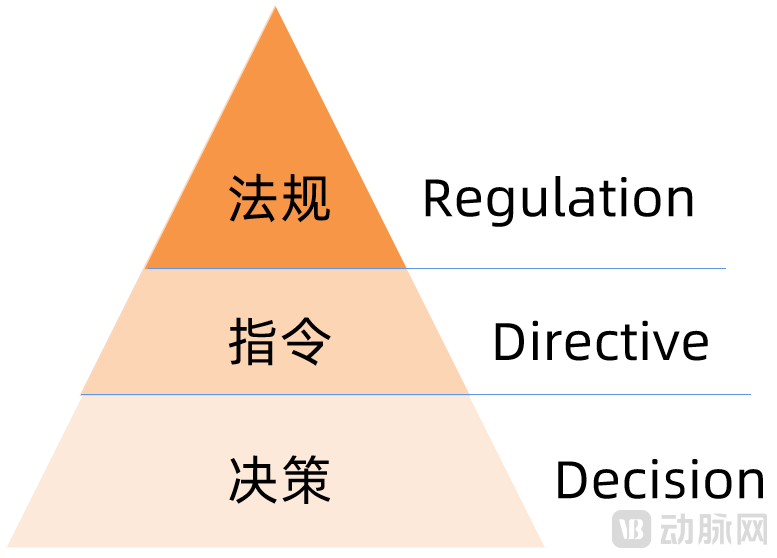
The Pyramid Structure of EU Regulations (in Descending Order of Binding Force)
From Directives to Regulations, the European Union has strengthened its regulatory oversight of medical devices. Regulations take immediate effect and become binding law across all EU member states upon publication, eliminating the need for transposition into national legislation as was required under the Directive framework.
It is worth noting that under the EU medical device classification system, medical devices are categorized into two major groups: Medical Devices (MD) and In Vitro Diagnostic Medical Devices (IVD). Currently, only Medical Devices are subject to the new Medical Device Regulation (MDR). The corresponding new regulation for in vitro diagnostics, the In Vitro Diagnostic Regulation (IVDR, EU 2017/746), came into effect on May 26, 2022. This means that IVD manufacturers still have one year to buffer and adapt to the regulatory changes and make adequate preparations for compliance.
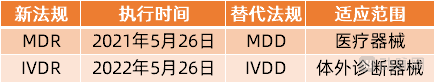
EU Medical Device Regulations for Two Classes of Devices and Their Implementation Timelines
For other medical device (MD) manufacturers, applications for EU certification submitted on or after May 26, 2021, must comply with the new Medical Device Regulation (MDR). What changes have been introduced under the new EU MDR compared to the previous Medical Device Directive (MDD)? In light of these changes, what key considerations should Chinese medical device manufacturers keep in mind when applying for CE certification? For manufacturers that have already obtained CE certification, what measures should they take to maintain their CE certificates in the future?
In the past, obtaining CE certification in the European Union was less challenging than securing approval from China’s National Medical Products Administration (NMPA) or the U.S. Food and Drug Administration (FDA). This disparity stemmed from the weaker enforceability of the EU’s former medical device regulations, which generally imposed lower submission requirements on manufacturers. Consequently, some medical devices that had only obtained CE certification were frequently associated with adverse events after being deployed in the EU market. As a result, these products will still face more prolonged and stringent regulatory review processes when seeking future market access in China and the United States.
Prior to the implementation of the new regulations, manufacturers of certain low-risk medical devices could apply for CE certification through self-declaration. However, this approach was subject to lax oversight and lacked enforceability. Consequently, in 2020, an exposé revealed that a medical device company based in Shenzhen, China, had exported products to the European Union via self-declaration, only for 90% of these products to be found non-compliant.
If a product cannot be precisely controlled, the CE certification carries significant “watered-down” credibility. According to industry insiders, in response to this situation within the EU, countries such as the United Kingdom and Germany have even established “short lists.” If a product has only obtained CE certification in the EU, it must still undergo rigorous clinical trials when applying for market approval in the UK and Germany, to ensure its safety and efficacy.
Changes in the Declaration Process for EU Medical Devices Under New and Old Regulations (Screenshot from Guanwu Xiaoer & the Official Website of the China Council for the Promotion of International Trade)
However, following the implementation of the EU’s new Medical Device Regulation (MDR), even medical products sold on e-commerce platforms are required to undergo certification through strictly authorized Notified Bodies (NBs). Concurrently, the EU has significantly raised its requirements for NBs. As third-party entities independent of the manufacturers whose products they assess for conformity, NBs are mandated to maintain permanent staff with relevant qualifications—such as product reviewers and quality management system auditors—and are prohibited from outsourcing these critical roles.
The stringent requirements for Notified Bodies (NBs) have also led to a significant reduction in the number of currently authorized bodies capable of issuing certifications under the new regulations. According to information available on the official European Union website, there were a total of 51 Notified Bodies previously designated under the Medical Devices Directive (MDD).
However, since May 26 this year, notified bodies designated under the MDD have been prohibited from issuing new certificates under that directive and are only permitted to oversee the transition of previously issued valid certificates. Statistics show that currently, only 20 public institutions are authorized under the EU’s new Medical Device Regulation (MDR).
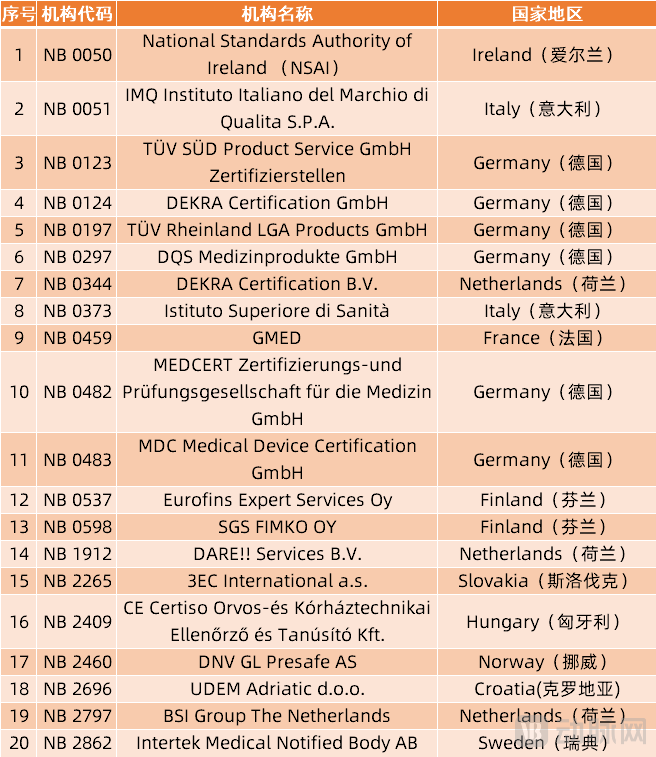
20 Notified Bodies Currently Authorized to Issue MDR Certificates (Source: European Union Official Website)
The sharp decline in the number of Notified Bodies (NBs) is one indication that EU certification has become more difficult. The new Medical Device Regulation (MDR) has implemented sweeping enhancements to clinical evaluation requirements. For instance, Article 15 of the MDR stipulates that medical device manufacturers must designate at least one person within their organizational structure responsible for regulatory compliance. This individual must have four years of professional experience in regulatory affairs or quality management systems related to medical devices; alternatively, they must hold a formal qualification in law, medicine, pharmacy, engineering, or another relevant discipline, along with at least one year of professional experience in medical device regulatory affairs or quality management systems.
With the raised thresholds for notified bodies and a significantly extended certification cycle, obtaining CE certification for medical devices in the EU under the new regulations will be no less challenging than securing FDA approval. VCBeat (WeChat ID: vcbeat) has identified several key directions of change under the new MDR regulations through a comparative study of the EU’s old and new frameworks:
1. Expansion of the scope of medical devices and refinement of medical device classification.
2. Establish a Unique Device Identification (UDI) system and database to strengthen device traceability management.
3. Heightened requirements for the safety, performance, and related documentation of medical devices.
Manufacturers are required to establish a post-market surveillance system for their devices and regularly update surveillance reports.
Under the new EU Medical Device Regulation (MDR), in addition to incorporating devices previously governed by the Medical Devices Directive (MDD) into the definition of medical devices, two additional categories of products for specific purposes have also been included:

EU MDR New Regulations Expand the Concept of “Medical Devices” (Screenshot from the Original MDR Text)
First, it clarifies that instruments and equipment used to obtain information through in vitro examination of human samples are included within the scope of medical devices. Second, it also brings under the regulatory purview of medical devices those instruments and equipment specifically designed for cleaning, disinfecting, or sterilizing other medical device products and active medical devices.
It is worth noting that the European Union distinguishes between in vitro diagnostics (IVD) and in vitro examinations. Devices related to in vitro diagnostics are subject to the In Vitro Diagnostic Regulation (IVDR, EU 2017/746), whereas devices related to in vitro examinations fall under the Medical Device Regulation (MDR, EU 2017/745).
Under EU regulations, medical devices are classified into four categories: Class I, Class IIa, Class IIb, and Class III. This risk-based classification is grounded in the vulnerability of the human body and takes into account the potential risks associated with the technical design and manufacture of the devices.
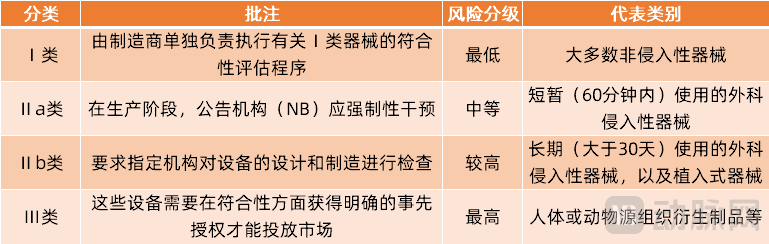
EU Medical Device Classification
Under the new EU MDR regulations, the basic classification of medical devices remains unchanged; however, the categories of specific products involved have been further refined, with particular improvements made to Class III medical devices. According to incomplete statistics, 12 new types of Class III medical devices have been added under the MDR, which are as follows:
1. Non-invasive substances that come into direct contact with human cells, tissues, or organs in vitro.
2. Medical devices specifically intended for direct contact with the central nervous system.
3. Implantable devices and long-term surgically invasive devices used for the management of pharmaceutical products.
4. Active implantable medical devices or their accessories.
5. Breast implants or surgical mesh.
6. Total or partial joint prostheses (excluding ancillary components such as screws, wedges, plates, etc.).
7. Intervertebral disc replacement implants or implantable devices in contact with the spine (as above, excluding auxiliary components).
8. All active devices intended to control, monitor, or directly affect the performance of active implantable medical devices.
9. Provide decision-making software for diagnostic or therapeutic purposes, where the decisions may lead to human death or irreversible deterioration of health conditions.
10. Devices composed of nanomaterials with high or moderate internal radiation exposure potential.
11. Devices composed of substances or combinations of substances that are introduced into the human body through bodily orifices or applied to the skin and are absorbed by the human body or locally dispersed (including cases where the substances and their metabolites are absorbed by the human body to achieve the intended purpose, or where the substances and their metabolites are absorbed in the stomach or digestive tract to achieve the intended purpose).
12. Active therapeutic devices with integrated or combined diagnostic functions, such as closed-loop systems or automated external defibrillators.

MDR: Refined Classification Compared to MDD
An analysis of the newly added provisions in the EU MDR reveals that the new regulation further refines the classification framework established under the previous Medical Devices Directive (MDD). It provides clear definitions for previously ambiguous categories and introduces a tiered classification system based on the varying severity of potential consequences associated with similar devices.
For example, in the classification of decision-making software, if its decisions may lead to human death or irreversible deterioration of health conditions, the software is classified as the most stringent Class III; if its decisions may cause significant deterioration in a person's health condition or surgical intervention, the software is categorized as Class IIb; and all other decision-making software intended for diagnostic, therapeutic, or physiological process monitoring purposes is classified as Class IIa.
Similarly, the MDR has also introduced new classification rules for devices that contain or are composed of nanomaterials: if the device presents a high or medium potential for internal exposure, it shall be classified as Class III; if there is a lower potential for internal exposure, it shall be classified as Class IIb; if the potential for internal exposure is negligible, it shall be classified as Class IIa.
In accordance with the principles of the EU classification system, devices or software that pose a greater potential impact on patients are assigned to more stringent regulatory classes. For instance, under Rule 20 of the Medical Device Regulation (MDR), all invasive devices intended for drug administration via inhalation through body orifices, excluding surgically invasive instruments, are classified as Class IIa. However, if the mode of action of such devices has a significant impact on the efficacy and safety of the administered medicinal product, or if they are intended to treat life-threatening diseases, they are classified as Class IIb.
Meanwhile, as the new Medical Device Regulation (MDR) redefines the types and availability of clinical data, it will also bring significant distinctions to the medical device risk classification for digital health applications (digital health APPs). Digital health applications (DiGA) that have already passed the German Federal Institute for Drugs and Medical Devices (BfArM) review need to confirm whether their product risk has been upgraded under the new MDR regulations. If an APP no longer meets the Class I or IIa requirements stipulated by the DiGA Act and is upgraded to Class IIb or even a higher risk classification, it will no longer qualify as a DiGA.
In addition to further refining the basic definitions and classifications of medical devices, another major improvement in the EU Medical Device Regulation (MDR) over the Medical Devices Directive (MDD) is the requirement to establish the European Database on Medical Devices (Eudamed) and the corresponding Unique Device Identification (UDI) system. This facilitates public access to comprehensive information on medical devices and enables traceability of each individual device.
The European Database on Medical Devices (Eudamed) shall comprise seven electronic systems: the device registration module, the UDI database, the economic operators registration module, the notified bodies and certificates module, the clinical investigations module, the vigilance and post-market surveillance module, and the market surveillance module.
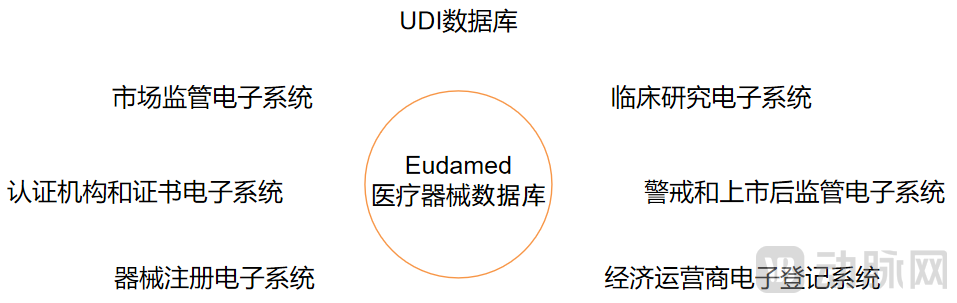
Components of the Medical Device Database (Graphic by VCBeat)
Data for these seven electronic systems are entered by Member States, notified bodies, economic operators, and sponsors. Eudamed compiles and processes all information, ultimately granting access to notified bodies, economic operators, sponsors, and the public. Specifically, for Class III devices and implantable devices (excluding custom-made or investigational devices), manufacturers must prepare a summary of safety and clinical performance, which shall be made publicly accessible through Eudamed.
It is worth noting that the UDI data required for entry into the medical device database serves as the “identity card” for medical devices, consisting of two components: the UDI-DI and the UDI-PI.

Composition of the Medical Device “ID Card” UDI (Graphic by VCBeat)
The UDI-DI can be understood as the “Product Identifier,” consisting of a company identifier and a product specification code. It is a fixed and mandatory component of the UDI, required to be uploaded to the UDI database for public access. In contrast, the UDI-PI serves as the “Production Identifier” of the device, comprising production-related information such as the medical device serial number, lot number, manufacturing date, and expiration date. It is not mandatory and does not need to be uploaded to the UDI database.
Globally, in 2008, the International Medical Device Regulators Forum (IMDRF) established a Unique Device Identification (UDI) Working Group, and in September 2011, it adopted the Guidance on the Unique Device Identification System for Medical Devices. In 2012, the IMDRF further supplemented and refined this framework by adopting the Proposed Rules for the Unique Device Identification System for Medical Devices, providing guidance to regulatory authorities worldwide in developing their own UDI systems in accordance with these proposed rules.
Currently, among the 10 countries and regions of the IMDRF, the United States and the European Union have already issued UDI-related regulations. However, in the U.S., FDA’s UDI system places responsibility on the labeler, whereas in the EU, it is the manufacturer that bears this responsibility, with specific requirements that labeling must be in the official language(s) of the EU member state(s). In addition, Brazil, China, and South Korea have successively released draft regulations for public comment, while Japan, Canada, and Australia have issued UDI-related notices and guidance documents. The era of comprehensive UDI implementation for medical devices is imminent!
Prior to the issuance of new EU regulations, it was relatively easier to obtain CE certification under the old EU Medical Device Directive (MDD). As a result, many domestic medical device manufacturers have secured CE certification for their products in overseas markets, yet they have still failed to obtain marketing authorization in China.
However, the mandatory enforcement of the new EU Medical Device Regulation (MDR) will significantly increase the difficulty of obtaining CE certification, while simultaneously enhancing its value and credibility. Under the new regulation, the European Union has emphasized the importance of improving the safety and performance of medical devices, as well as strengthening documentation requirements, and has reinforced the post-market surveillance system for these devices.
Under Chapter I, General Requirements, of Annex I to the MDR, manufacturers are required to establish, implement, document, and maintain a risk management system. Risk management shall be understood as a continuous iterative process throughout the entire lifecycle of the device, requiring regular systematic updates. Furthermore, manufacturers must draft a Summary of Safety and Clinical Performance (SSCP) covering eight aspects, including the UDI-DI, submit it to the CE Notified Body for verification, and subsequently upload the summary report to the Eudamed database for regulatory oversight.
Furthermore, the MDR has added requirements for the content of technical documentation and explicitly states that the post-market surveillance plan and the Periodic Safety Update Report (PSUR) are part of the technical documentation. It also requires that relevant information in the technical documentation be updated based on data collected through the post-market surveillance system.
Medical device manufacturers shall plan, establish, document, implement, maintain, and update a post-market surveillance system. The post-market surveillance system shall be suitable for the active and systematic collection, recording, and analysis of quality, performance, and safety-related data on the device throughout its lifecycle, in order to draw necessary conclusions and identify, implement, and monitor any preventive and corrective actions.
For the PSUR, manufacturers of Class IIa, IIb, and III devices shall prepare Periodic Safety Update Reports (PSURs) for each device, category, or group of devices, summarizing the results and conclusions of the data analysis, and providing rationale and explanations for any preventive and corrective actions taken. Among these, manufacturers of Class IIb and III devices shall update the PSUR at least annually; manufacturers of Class IIa devices shall update the PSUR at least every two years, as necessary.
Finally, the EU’s new Medical Device Regulation (MDR) has also enriched the clinical evaluation component of medical device audits by establishing dedicated expert panels to assess and review the intended clinical purpose and clinical investigation plans of medical devices submitted by manufacturers for approval. Manufacturers shall give due consideration to the opinions issued by these expert panels but shall not exercise any rights over the views expressed by the panels regarding any future conformity assessment procedures.
An analysis of the EU’s new Medical Device Regulation (MDR) reveals that its enhancements have, to some extent, drawn upon established international medical device regulations, particularly the U.S. Food and Drug Administration’s (FDA) approval requirements for medical devices. This alignment has further advanced global consensus on standardized medical device regulations.
“Strict regulations have an overall positive impact on the healthy development of the entire healthcare industry,” affirmed Yan Jing, CEO of Huixian Medical, a company specializing in R&D of upstream medical devices for in vitro diagnostics. “Although the implementation of the EU’s new Medical Device Regulation (MDR) will pose challenges for Chinese exporters—such as increased costs, prolonged certification cycles, and heightened compliance risks—the regulation encourages medical device manufacturers to adhere to more stringent standards and ensure product and regulatory compliance. This is ultimately beneficial to patients and represents both a challenge and an opportunity for the entire medical device industry.”
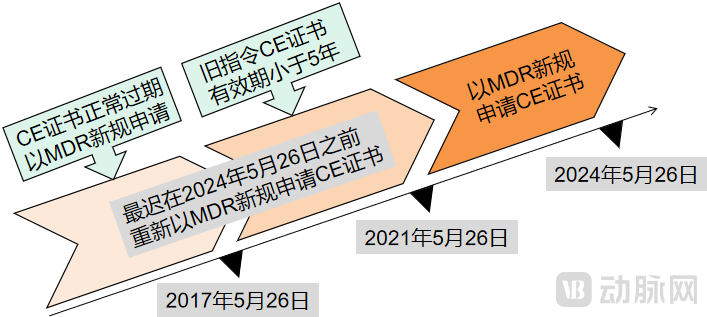
Key Considerations for Companies That Obtained CE Certificates at Different Times(Chart by VCBeat)
CE certificates issued by notified bodies under the previous Directives (MDD, AIMDD, or IVDD) during the transition period shall remain valid for their duration, provided that such validity does not exceed five years from the date of issuance and expires no later than 26 May 2024.
For medical device products that have already been placed on the market with CE certificates obtained prior to the transition period, manufacturers must reapply for CE certification under the new Medical Device Regulation (MDR) as soon as possible, and no later than May 26, 2021, before the expiration or invalidation of the old CE certificate. This process involves confirming whether the product’s risk classification has been upgraded, verifying whether the original notifying body that issued the CE certificate still holds MDR designation status, promptly updating the original CE technical documentation, and submitting a new certification application to a Notified Body (NB) authorized under the MDR.
"This means that after 2024, there will no longer be any CE certificates based on the old regulations in the entire EU market. It is urgent for medical device developers to adapt to the new regulations as soon as possible. 'Medical device developers who prepare early for the new regulations will see increased revenue in the future, while 20%-30% of small and medium-sized medical device manufacturers in the EU will face elimination,' said Zhang Jing, CEO of Zhisu Health, a 3D-printed medical device company."
Meanwhile, due to the heightened entry requirements in the European Union and stricter regulations on the research and development process of medical devices, the traditional domestic pathway of OEM contract manufacturing followed by CE certification in the EU will no longer be viable. Companies whose core business is contract manufacturing of medical devices will be forced to transform. Nevertheless, this development is undoubtedly a significant boon for the overall growth of the EU medical device market. As the second-largest medical device market globally after the United States, accounting for 26% of the global total, maintaining market access for medical device companies is crucial.