China's NMPA Unveils New Regulatory Framework for AI Medical Software, Clarifying Classification and Approval Pathways
On July 8, the National Medical Products Administration (NMPA) released on its official website the “Notice of the National Medical Products Administration on Issuing the Guiding Principles for the Classification and Determination of Artificial Intelligence Medical Software Products (No. 47 of 2021),” formally issuing the “Guiding Principles for the Classification and Determination of Artificial Intelligence Medical Software Products” (hereinafter referred to as the “Guiding Principles”), which clarifies the classification and determination of artificial intelligence medical software products.
Coincidentally, July 8 also marked the opening day of the 2021 World Artificial Intelligence Conference, a grand event in the AI field, with leading figures from the medical AI industry gathering in Shanghai. Upon learning this news, it may well evoke “mixed feelings—joy for some, sorrow for others.”
So, what changes have been made in the “Guidelines” compared to previous regulations, and what impact might they have on the industry? VCBeat (WeChat ID: Vcbeat) provides an analysis.
With technological advancements, software products leveraging artificial intelligence (AI) have been increasingly widely adopted in the healthcare sector. As early as 2017, the National Medical Products Administration (NMPA) established an AI Working Group within its subordinate technical department to conduct research on the regulation of AI-based medical devices. Key research areas include the establishment of evaluation databases for AI medical devices, algorithm assessment, and data security, with an emphasis on integrating industry, academia, research, medical practice, and inspection, as well as fostering interdisciplinary collaboration.
Nevertheless, as is typical in the early stages of emerging technology applications, regulatory frameworks for medical artificial intelligence (AI) software require time to be further refined. In particular, the classification and definition of AI software were previously ambiguous, encompassing both scenarios where such software is regulated as a medical device and where it is not. Meanwhile, there have been numerous instances of “regulatory gray-area” practices in the approval of registration certificates.
For example, prior to 2020, although some medical device products submitted for registration incorporated deep learning algorithms as aids, the term “artificial intelligence” was not used as a key element in their regulatory submissions. Therefore, even though these Class III medical devices were supported by algorithms such as deep learning and machine learning, they were approved through standard regulatory pathways and cannot be classified as AI-based medical software.
At that time, the prolonged failure to secure regulatory approvals undoubtedly posed a significant headwind to the development of medical artificial intelligence. After all, as early as the beginning of 2018, quiet debates had already begun regarding the timeline for the approval of the first medical AI product. However, with no medical AI products gaining approval over an extended period, market patience was gradually being exhausted.
Based on extensive research and exchanges with relevant R&D enterprises, universities, hospitals, research institutions, and overseas regulatory authorities, the National Medical Products Administration (NMPA) issued the “Review Points for Deep Learning-Aided Decision-Making Medical Device Software” in July 2019.
This document covers the scope of application, key points for regulatory review, software updates, relevant technical considerations, and instructions for registration submission materials. It clarifies the fundamental considerations for evaluating the safety and effectiveness of artificial intelligence products based on deep learning for assisted decision-making, thereby guiding the research and development as well as registration submissions of such products.
Meanwhile, in 2019, the National Medical Products Administration (NMPA), in collaboration with relevant national departments, research institutions, academic institutes, medical institutions, and professional societies, jointly established the Artificial Intelligence Medical Device Innovation Cooperation Platform. The platform set up ten working groups, including the Technical Regulations Working Group, Data Governance Working Group, Evaluation Database Construction Working Group, Cybersecurity Working Group, Standardization Research Working Group, Evaluation Technology Research Working Group, Clinical Evaluation Working Group, Real-World Data Application Working Group, Talent Development Working Group, and International Exchange Working Group. Through this cooperation platform, the working groups are encouraged to conduct multi-level and multi-perspective scientific research, comprehensively utilize various information technology tools, promote technical integration across the entire industry chain, and facilitate the international development of artificial intelligence technologies and industries.
For instance, China has taken a leading role internationally in the standardization of artificial intelligence (AI) medical software. In December 2018, China spearheaded the establishment of the IEEE Artificial Intelligence Medical Device Working Group (AIMDWG) under the Institute of Electrical and Electronics Engineers (IEEE). Two proposals for IEEE international standards were submitted and approved. Currently, international standard research is being conducted in two key areas: evaluation terminology for the effectiveness and safety of AI medical devices, and quality management and evaluation of datasets. This marks the first time that China has led the development of international standards in the field of AI medical devices.
In 2019, the national technical focal point for artificial intelligence medical device standardization was established. In 2020, two foundational industry standards—“Quality Requirements and Evaluation of Artificial Intelligence Medical Devices—Part 1: Terminology” and “Quality Requirements and Evaluation of Artificial Intelligence Medical Devices—Part 2: General Requirements for Datasets”—were approved for development. The drafting of these standards has been completed, and they have been released for public comment within the industry, laying a solid foundation for standardization efforts.
The long-awaited turning point in the approval of artificial intelligence (AI) medical software finally arrived in 2020. On January 15, 2020, the National Medical Products Administration (NMPA) issued the first medical device registration certificate for an “AI”-based product. Keya Medical’s coronary flow reserve fraction calculation software was the first to cross the finish line, marking a new stage in the commercialization of medical AI.
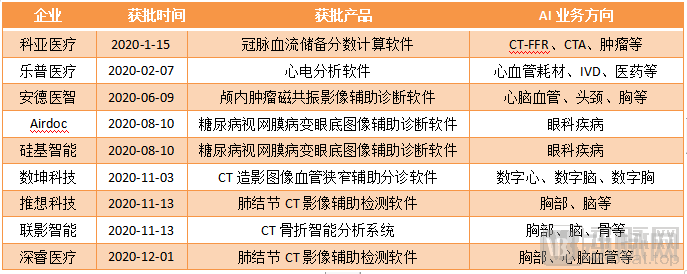
Medical AI Approved by the National Medical Products Administration in 2020
Subsequently, the approval process accelerated sharply. In 2020 alone, nine “AI” medical device registration certificates were approved. In 2021, additional “AI” registration certificates for companies such as Deepwise Healthcare were successively granted. According to statistics, from 2021 to the present, the National Medical Products Administration (NMPA) has cumulatively approved 15 “AI” medical device registration certificates.
Nevertheless, the details of regulatory rules remain to be refined. In early 2020, during interviews conducted by VCBeat with relevant approved enterprises, industry insiders noted that the “Review Points for Deep Learning-Assisted Decision-Making Medical Device Software,” which later served as the primary basis for medical device certification, had not yet been released during the product development phase. This absence left the R&D process without authoritative review guidelines or comparable products for reference, resulting in some detours. Therefore, it is urgent to further improve the regulatory framework.
In 2021, to standardize the attribute definition and classification of artificial intelligence (AI)-based medical software, and to provide technical guidance on registration and approval for the industry and regulatory authorities, the Medical Software Specialist Group of the Medical Device Classification Technical Committee of the National Medical Products Administration (NMPA) developed the “Guiding Principles for the Classification and Definition of AI-Based Medical Software.” This development was based on a review of the current status of registered AI-based medical software products both domestically and internationally, as well as relevant classification policies for medical software products at home and abroad. It also took into account the technical characteristics of AI algorithms and the level of risk associated with their regulation, in accordance with the Regulations on the Supervision and Administration of Medical Devices, the Rules for Medical Device Classification (Order No. 15 of the China Food and Drug Administration), and the Medical Device Classification Catalog.
On April 16, 2021, the National Medical Products Administration released the “Guiding Principles for the Classification and Identification of Artificial Intelligence-Based Medical Software Products (Draft for Comments).” After a period of public consultation, the Guiding Principles have finally been issued.
So, what are the differences between the “Guiding Principles” and the “Draft for Comments”? The differences are mainly reflected in four aspects: scope, definition of regulatory attributes, classification of regulatory categories, and implementation requirements.
Integrated hardware and software solution included
First, there are certain differences between the two in terms of the scope of AI-based medical software.
In the “Draft for Comments,” the scope is only briefly mentioned: “AI-based medical software refers to standalone software that employs artificial intelligence (AI) technology to fulfill its intended use in the medical field.”
However, the official “Guiding Principles” describe the scope as follows: “AI medical software refers to standalone software that utilizes artificial intelligence technologies to achieve its intended medical purposes based on medical device data. The classification and determination of medical devices containing AI software components may refer to these Principles.” Additionally, it specifically defines “medical device data” as objective data generated by medical devices for medical purposes; under special circumstances, this may include objective data generated by general-purpose equipment for medical purposes.
In contrast, the official version of the Guidelines is more rigorous and encompasses integrated hardware-software solutions. Indeed, such integrated solutions are among the mainstream approaches.
More Precise Scope of AI Medical Software
In terms of attribute definition, the Draft for Comments states that “software employing artificial intelligence algorithms, whose intended use conforms to the definition of medical devices as stipulated in Article 76 of the Regulations on the Supervision and Administration of Medical Devices, shall be regulated as medical devices.” Furthermore, AI-based medical software is categorized into four types according to its intended use: decision support software, imaging/data processing software, analysis and mining products, and medical assistant products.
Among these, decision support software and imaging/data processing software are explicitly regulated as medical devices, whereas analysis and data mining products and medical assistant products do not fall under the category of medical devices.
The description in the official version of the "Guiding Principles" has subtly changed, emphasizing that "the determination of a product's regulatory classification should be based on its intended use, with comprehensive judgment made in conjunction with factors such as the target of processing and core functionality."
Specifically, if the processing object of a software product is medical device data, and its core functions involve the processing, measurement, model calculation, and analysis of such data for medical purposes, it shall meet the definition of a medical device under the Regulations on the Supervision and Administration of Medical Devices and be regulated as a medical device.
Conversely, if a software product processes non-medical device data (such as patient chief complaints and other information, or conclusions from laboratory and imaging reports), or if its core functions do not involve the processing, measurement, model-based calculation, or analysis of medical device data, or if it is not intended for medical purposes, it shall not be regulated as a medical device.
In contrast, the final version of the “Guiding Principles” adopts more consistent principles for determining the regulatory status of medical device software, specifically by defining it based on whether the data processed constitutes medical device data and the intended purpose of such processing, thereby ensuring greater rigor. Meanwhile, it avoids the more absolute wording found in the “Draft for Comments” and prevents a rigid perception of classification.
For instance, analytical and data mining products, as well as medical assistant products, which were explicitly excluded from the definition of medical devices in the Draft for Comments, may in practice fall under medical device regulation due to differences in their intended use and processing methods. The final version of the Guidelines closes this regulatory gap by bringing such products within the scope of regulation.
Clear Path to Class II Certification Approval
“The Draft for Comments” defines the classification category based on whether a diagnostic conclusion is provided. Medical software that does not employ artificial intelligence technology and is used solely for image/data processing, without providing diagnostic conclusions or performing drug calculations, is regulated as a Class II medical device. Medical software used to provide diagnostic conclusions or perform drug calculations is regulated as a Class III medical device.
Given the poor interpretability of artificial intelligence technologies, their application in the medical field is still in its infancy, and the clinical practical risks have not yet been comprehensively and thoroughly evaluated, the "Draft for Comments" holds that, in principle, medical software employing artificial intelligence technologies should currently be regulated as Class III medical devices.
The official version of the Guidelines introduces a critical shift, determining the regulatory classification primarily based on factors such as the product’s intended use and algorithm maturity. According to interviews conducted by VCBeat with industry experts, this will provide the fundamental basis for product classification both currently and in the future.
Specifically, AI-based medical software with low maturity in healthcare applications (i.e., not yet marketed or with insufficiently demonstrated safety and effectiveness) shall be regulated as Class III medical devices if used for clinical decision support, such as providing recommendations for lesion feature recognition, determination of lesion nature, medication guidance, and formulation of treatment plans.
If used for non-assistive decision-making purposes, such as providing clinical reference information through data processing and measurement, it shall be regulated as a Class II medical device.
For AI-based medical software with a high level of algorithmic maturity in clinical applications (i.e., safety and effectiveness have been fully demonstrated), the classification shall be determined in accordance with the current “Medical Device Classification Catalog” and related classification determination documents.
According to the definition in the “Draft for Comments,” AI-based medical software will basically be regulated as Class III medical devices. Although it is further clarified that the classification category may be adjusted later based on the clinical risks of AI-based medical software and the outcomes of their technical evaluation, such a provision is virtually negligible in practical implementation.
However, the final version of the “Guiding Principles” clarifies that AI-based medical software intended for non-assistive decision-making will be regulated as Class II medical devices. Given that Class II certifications can be approved by local authorities, they are easier to obtain. Industry experts note that the new regulations open up possibilities for classifying post-processing products as Class II devices, demonstrating support from relevant authorities for industry development.
Transition period until the end of 2023
There are notable differences in implementation requirements between the Exposure Draft and the final version of the Guidelines. The Exposure Draft merely states that it shall come into force on the date of promulgation and implementation, without any further elaboration.
In the official version of the Guidelines, different definitions are provided in light of current practical circumstances. It is clarified that products already approved or under application shall continue to be governed by the original regulations and remain unaffected. Meanwhile, it is also stipulated that existing registration certificates shall, in principle, expire no later than December 31, 2023.
Industry experts believe that this will have no impact on the three Class III products currently approved, but it will have a certain impact on some Class II AI software products previously approved by local authorities, which will need to be prepared for submission for registration under the new management category. If an upgrade to Class III is required, the original registration certificate can be extended until the end of 2023. In addition, there is also the possibility that approval may not be granted under the new regulations after the reclassification.
Medical devices represent one of the most dynamically innovative sectors within the broader health industry. Both the National 13th Five-Year Plan for Scientific and Technological Innovation and the National Strategy for Innovation-Driven Development explicitly prioritize the research and development of high-performance medical devices. In 2015, the State Council issued the Opinions on Reforming the Review and Approval System for Drugs and Medical Devices, and in 2017, the General Office of the Communist Party of China Central Committee and the General Office of the State Council jointly issued the Opinions on Deepening the Reform of the Review and Approval System to Encourage Innovation in Drugs and Medical Devices. Both documents clearly outlined requirements and roadmaps for promoting industrial innovation and development.
With the release of these Guidelines, the definition, classification, and regulation of AI-based medical software products have become clearer and more operational, providing explicit guidance for companies in the industry on product positioning and registration strategies. Furthermore, downgrading certain AI medical software products to Class II management will also promote industry development. The policy tone supporting and encouraging the innovative development of China’s medical device products, including AI-based products, is now clearly evident.
Meanwhile, the National Medical Products Administration (NMPA) has been closely monitoring and tracking the regulatory approaches of advanced overseas regulatory authorities toward artificial intelligence (AI) products. The NMPA has stated that it will continue to encourage and support innovation in AI products while further refining and detailing review and approval requirements in light of domestic product submission trends, thereby facilitating the early market launch and application of such products. In early 2021, the NMPA issued a call for proposals for standard-setting projects in the field of AI medical devices to be initiated in 2022.
VCBeat will continue to closely monitor developments in regulatory oversight and the refinement of standards in this area. Stay tuned.