Sharpening the Blade: How Single-Cell Sequencing Technology Fights Disease
Editor’s Note: This article is republished from Hillhouse Capital, with authorization granted to VCBeat.
From Mendel’s discovery in his pea fields that genes determine biological traits, to the later revelation that nucleic acid sequences encode life, and further to advancements in biological cloning and gene editing, the development of genetic technology has gradually clarified the blueprint of life.
We all develop from the fusion of sperm and egg, yet the cells in our bodies differ markedly: some provide structural support, while others protect the organism; despite sharing identical DNA, they exhibit distinct morphologies and functions.
All of this stems from the complexity of differential gene expression across different cell types. Although all our somatic cells contain essentially the same genome, not all genes are expressed. This is akin to how most people do not use knowledge of conic sections after high school graduation, despite having learned it. By detecting nucleic acid sequences within cells, we can determine which genes are being expressed and thereby infer the specific functions or tasks the cells are performing.
Single-cell sequencing, as a tool capable of efficiently detecting heterogeneous gene expression across different cells, has been propelled to the forefront of our era, becoming an indispensable and powerful technology in the field of life sciences and medicine. This technology also enables us to analyze the differences and origins of the diverse cell types within our bodies, thereby aiding us in combating diseases.
Cellular characteristics are determined by the DNA they possess. Multicellular organisms arise from the replication and differentiation of a single fertilized egg, meaning that all cells within an individual share the same DNA. Despite having highly similar DNA, cells exhibit distinct differences, including cancer cells. Although cancer cells share the same set of genetic information as other somatic cells, their behavior is entirely contrary to what our bodies require.
How to Detect Cancer Cells Hidden in the Body Has Become a Major Challenge in Medicine.
The differences between cancer cells and other normal cells are minimal; a single-point mutation in one gene, or even a slight overexpression of a protein, can push a cell down the path of no return. These subtle yet critical mutations require meticulous identification and screening. If we sequence large tissue samples all at once, these minor discrepancies may be dismissed as errors, causing us to miss the detection of cancer cells.
Therefore, individual cell screening is essential for cancer treatment.
Direct protein analysis of individual cells is challenging. The extremely complex structure of cellular proteins and the difficulty in isolating them make it impossible to accurately analyze the proteome of a single cell. Rather than standing at the door of a cake shop counting every customer who buys a cake, it is far more efficient to consult the ledger. Similarly, scientists have turned their attention to mRNA, the “ledger” that determines protein expression.
mRNA serves as the messenger of gene expression; when a gene needs to be translated into protein, the corresponding mRNA is first transcribed. To determine which cakes are bestsellers in a bakery, one can simply analyze the number of printed orders from the kitchen. Similarly, by analyzing the types and quantities of mRNA, we can assess the operational status of a cell. Compared to proteins, mRNA signals can be precisely amplified, enabling single-cell RNA sequencing technology to more readily identify the types and quantities of proteins expressed by a cell, thereby determining its potential functions.
In addition to tumors, single-cell RNA sequencing technology can also detect mRNA expression profiles of different cells during embryonic development, thereby identifying key genes at various stages of organ development and providing a reference for stem cell-based organ transplantation.
Additionally, we can directly sequence common DNA mutation sites in cancer cells to analyze whether these key DNA sequences carry potential risks of variation, thereby assessing the status of the cells.
Currently, single-cell sequencing has been applied to a wide range of biomedical research and clinical trials, including tumor detection, embryonic development, immune cell therapy, and stem cell differentiation, making significant contributions to humanity’s exploration of the secrets of life and its fight against disease.
The workflow for single-cell sequencing is straightforward, comprising just three steps: nucleic acid extraction, library construction, and sequencing. For mRNA sequencing, reverse transcription of mRNA into cDNA is generally required to enhance sequencing stability.
For single-cell sequencing, we only need to isolate each cell and then sequence them individually.
Let us analyze this sequencing approach step by step: individual cells are isolated using techniques such as fluorescence-activated cell sorting (FACS) or laser capture microdissection (LCM), followed by nucleic acid extraction. However, this method is akin to husking rice grains one by one; given that a tissue sample typically contains tens of thousands of cells, such an approach is clearly inadequate and inefficient.
The clever application of barcodes has improved the efficiency of single-cell identification: instead of isolating and extracting individual cells one by one, we can simply tag each cell with a unique nucleic acid sequence barcode prior to sequencing and analysis.
This strategy is akin to a bank’s queue management system: customers are assigned different tags and directed to specific windows for queuing as needed. Countless "lobby manager" hydrogels distribute distinct "nucleic acid tag" numbers to different customers and guide them one by one into the hall to process "sequencing" services. A single experiment can capture information from hundreds to thousands of individual cells.
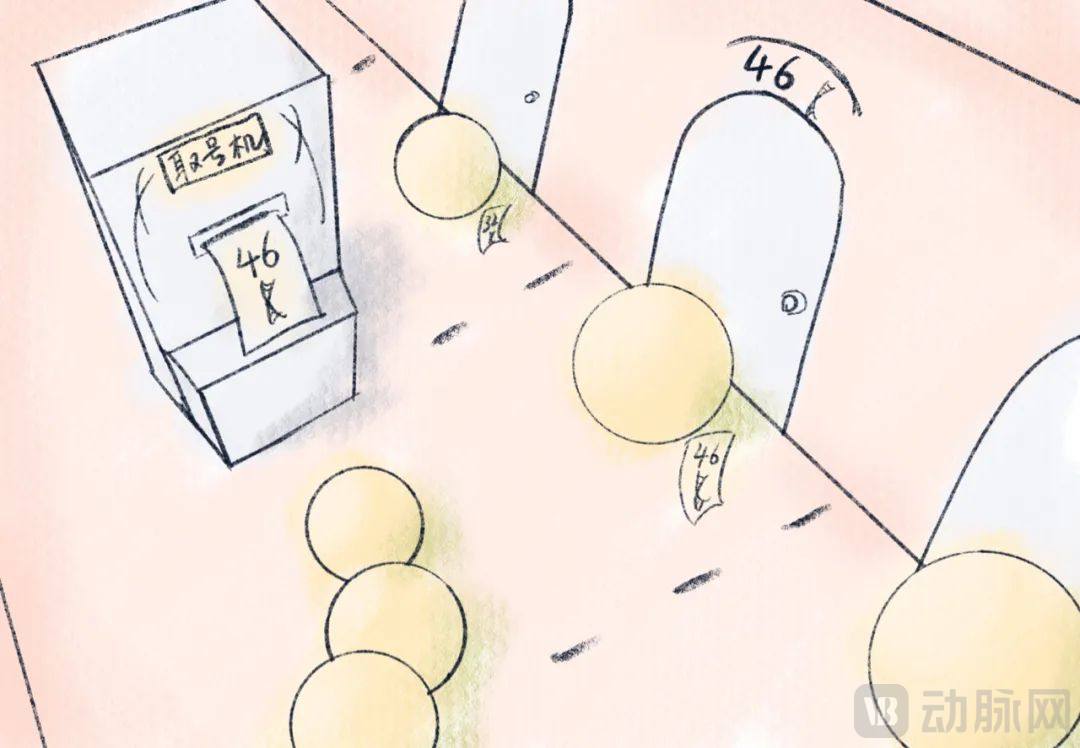
Schematic Diagram of Single-Cell Isolation | Illustrator: Yueyang Wudao
Furthermore, validation of single-cell sequencing results is essential. Commonly used methods include bulk (pooled) sequencing and proteomic analysis. Bulk sequencing involves direct whole-tissue sequencing and serves as a complementary approach to single-cell sequencing. Analogous to blending all fruits together to produce a mixed juice, bulk sequencing enables comparison with single-cell sequencing data to assess the relationship between single-cell findings and the overall tissue profile, thereby facilitating the precise definition of “abnormalities.” Proteomics directly analyzes the expression of key proteins within cell populations, providing corroboration for mRNA sequencing data.
The earliest gene sequencing technologies were based on the DNA replication process, converting DNA sequence information into readable signals.
The process of gene replication is akin to assembling LEGO bricks: using one original DNA strand as a template, DNA polymerase joins nucleotides together one by one according to the principle of complementary base pairing.
First-Generation Sequencing (Sanger Method) utilizes DNA polymerase synthesis reactions (PCR), incorporating a small amount of radioisotope-labeled ddNTPs into the standard PCR reaction system to determine a complete DNA sequence.
The unique structure of ddNTPs terminates the PCR reaction, much like a LEGO brick without studs prevents subsequent bricks from attaching, thereby halting chain elongation. Since ddNTP incorporation occurs randomly during PCR, termination can happen at any position. By ensuring that synthesis termination occurs at least once at every site where a specific base is present, each termination point corresponds to the location of that base. Finally, through gel electrophoresis and autoradiography, we can determine the bases at all termination sites, thereby obtaining the complete DNA sequence.
First-generation sequencing is the simplest and offers high accuracy, but it can only sequence limited lengths at a time. Much like the game Snake, where the snake becomes increasingly difficult to control as it grows longer, first-generation sequencing becomes less stable with increasing read length. The maximum read length for first-generation sequencing is approximately 1,000 base pairs (bp), making it unsuitable for longer sequences and entirely inadequate for whole-genome sequencing involving hundreds of millions of bases.
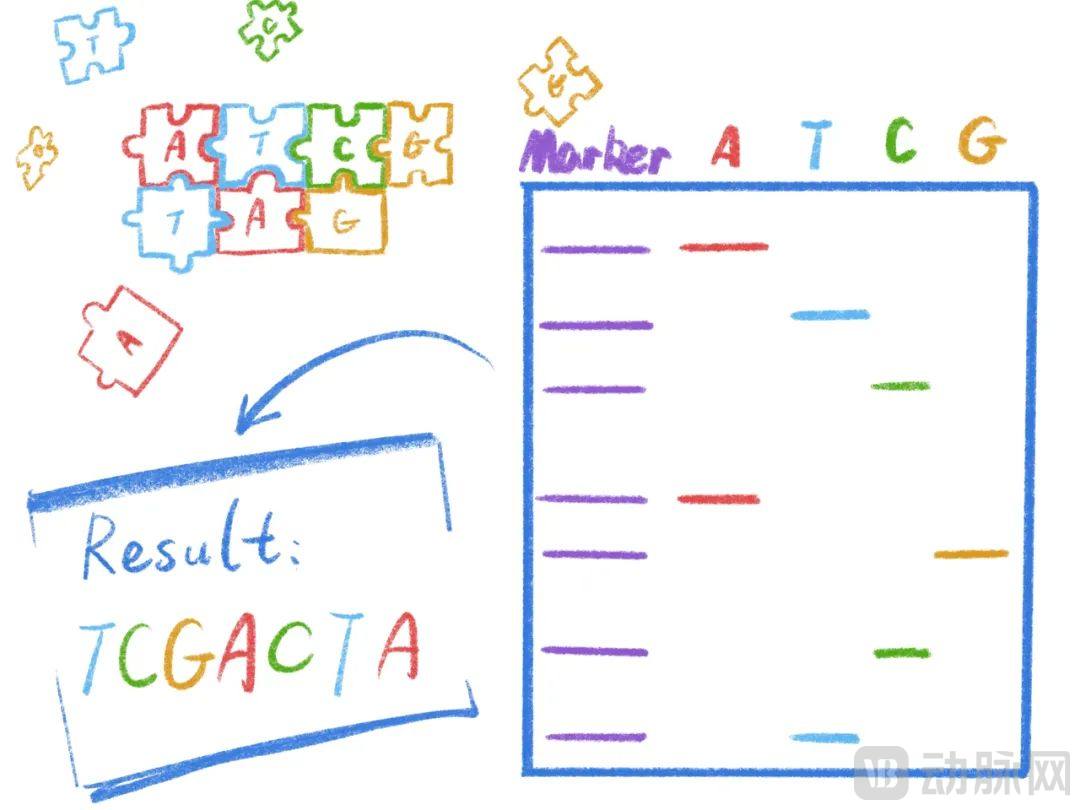
Principles of First-Generation Sequencing | Illustrator: Kirishima Tsukihi
Since we cannot sequence the entire genome in one go, we can sequence parts of it and then assemble them together!
Next-generation sequencing (NGS) is based on the “shotgun” approach, which fragments lengthy genomes into smaller pieces, sequences these fragments individually, and then assembles them into a complete sequence. Since comprehensive DNA reference libraries for common species have already been established, in clinical diagnostics and treatment applications, we only need to align a short DNA segment with the database to identify the original DNA within this library. The entire process is akin to a search operation, rather than piecing together associated sequence fragments one by one as in solving a jigsaw puzzle.
Although second-generation sequencing still relies on PCR-based DNA amplification, it employs the principle of "sequencing by synthesis," whereby the four nucleotide bases generate distinct fluorescent signals during DNA synthesis; these signals are captured in real time by high-precision optical cameras, enabling direct and immediate base calling.
Next-generation sequencing has significantly increased sequencing throughput, reduced sequencing costs and turnaround time, and features a high degree of automation, making it the most prominent sequencing technology in the current market.
Second-generation sequencing still has its own drawbacks: the fluorescence signal from individual bases is weak, making optical instruments prone to reading errors; sequencing fragments require replication and amplification, during which base mismatches and losses may occasionally occur, with these errors accumulating as the sample size increases; the read length per sequencing run is relatively short, leading to potential mismatches during alignment with reference databases.
Due to these technical limitations, there is a growing demand for technologies that do not require amplification and offer longer read lengths.
Third-generation sequencing retains the high-throughput characteristics of second-generation sequencing while employing single-molecule reading technology. This approach eliminates the need for PCR amplification, effectively reducing base information loss and mismatches caused by amplification, and increases read lengths to approximately 10 kb.
Currently, both second- and third-generation sequencing technologies rely on capturing weak fluorescence signals, necessitating the use of expensive optical monitoring systems. Furthermore, their reliance on the principle of DNA replication makes them heavily dependent on the activity of DNA polymerase. These characteristics significantly increase the cost of sequencing.
Fourth-generation sequencing technologies aim to perform sequencing without the need for DNA synthesis.
Fourth-generation sequencing enables real-time sequencing by guiding target sequences through a nanopore via primers and reading the distinct electrical signal variations as different bases pass through. This technology not only reduces instrument and consumable costs while achieving high accuracy, but also significantly miniaturizes the device footprint, shrinking sequencers from desktop-sized units to the size of a USB flash drive. The first domestically produced fourth-generation sequencer, developed by Qitan Technology, provides technical support for the widespread clinical application of single-cell sequencing.
In 2009, the first single-cell transcriptome sequencing technology, mRNA-Seq, ushered in a pivotal era for single-cell sequencing in the field of biomedicine. In 2014, single-cell sequencing technology was named “Method of the Year” by the prestigious international academic journal Nature Methods. In the same year, Science magazine ranked single-cell sequencing as the top breakthrough to watch, acknowledging that this technology has brought unprecedented changes to the biological and medical communities.
In 2017, the Human Cell Atlas (HCA) project was launched, a venture comparable in scale and ambition to the Human Genome Project. The HCA is a large-scale international collaborative initiative aimed at functionally classifying and defining all human cell types based on their unique biomolecular profiles. A critically important technology underpinning this endeavor is single-cell RNA sequencing (scRNA-seq). scRNA-seq can elucidate the gene structure and gene expression status of individual cells, thereby highlighting cellular heterogeneity. Its applications in the field of life sciences and medicine are becoming increasingly widespread.
In the near future, single-cell sequencing technology will expand the horizons of precision medicine, enabling more patients to access affordable, highly specific, and effective therapies, thereby improving treatment efficacy and the overall patient experience.
Single-cell RNA sequencing, a pioneering technology of our time, has become an indispensable tool in the field of life sciences and medicine. It currently plays a crucial role in oncology, developmental biology, microbiology, neuroscience, and other disciplines. With continuous technological advancements and breakthroughs, single-cell sequencing is poised to have broad application prospects in precision disease research and therapy, shining even more brightly on the stage of human health.
GL Ventures is Hillhouse’s venture capital fund dedicated to early-stage innovative companies, with a focus on key sectors including hard technology, software, biotechnology, new materials, and emerging consumer brands. GL Ventures seeks out entrepreneurs who are passionate about technology and believe in innovation. We aim to be the first call for founders seeking financing, and we look forward to accompanying them throughout their entrepreneurial journey over the long term.
References:
1. Sanger F, Nicklen S, Coulson AR. DNA sequencing with chain-terminating inhibitors. Proc Natl Acad Sci U S A. 1977;74(12):5463-5467. doi:10.1073/pnas.74.12.5463
2. MardisE R. Next-Generation DNA Sequencing Methods[J]. Annual Review of Genomics &Human Genetics, 2008, 9(9): 387-402
3. Van Dijk EL, Jaszczyszyn Y, Naquin D, Thermes C. The Third Revolution in Sequencing Technology. Trends Genet. 2018 Sep;34(9):666-681.
4. Stark R, Grzelak M, Hadfield J. RNA sequencing: the teenage years. Nat Rev Genet. 2019 Nov;20(11):631-656.
5. Potter SS. Single-cell RNA sequencing for the study of development, physiology and disease. Nat Rev Nephrol. 2018 Aug;14(8):479-492.6. Svensson V, Vento-Tormo R, Teichmann SA. Exponential scaling of single-cell RNA-seq in the past decade[J]. Nature Protocols, 2018, 13(4):599.7. Kolodziejczyk AA, Kim JK, Svensson V, Marioni JC, Teichmann SA. The technology and biology of single-cell RNA sequencing. Mol Cell. 2015 May 21;58(4):610-20.8. Method of the Year 2013. Nat Methods 11, 1 (2014).9. Tang F, Barbacioru C, Wang Y, et al. mRNA-Seq whole-transcriptome analysis of a single cell.[J]. Nature Methods, 2009, 6(5):377-382.10. Biological Encyclopedia. Dual Power: Joint Analysis of scRNA-seq and Bulk RNA-seq. Zhihu. https://zhuanlan.zhihu.com/p/334309279