Gene Therapy for Monogenic Rare Diseases: A One-Time Cure Approach
September 10–12, 2021: The 10th China Rare Disease Summit Held in Hangzhou. Themed “Walking Together on the Rare Disease Journey: A Decade of Shared Commitment,” the summit was jointly hosted by the Center for Rare Diseases (CORD), Zhejiang University School of Medicine, and the Second Affiliated Hospital of Zhejiang University School of Medicine.
More than 800 attendees from diverse sectors of the rare disease ecosystem—including government, industry, academia, and research, both domestic and international—convened for a three-day forum centered on the “patient-centric” philosophy. They engaged in multidimensional discussions to examine the development and opportunities within the rare disease industry. The forum was simultaneously live-streamed on three platforms: CCMTV Clinical Channel, Sina Health, and Wei Jie Yao, accumulating over 800,000 views.

At the opening ceremony of the forum, Ke Yuehai, Vice Dean of Zhejiang University School of Medicine; Wu Zhiying, Vice President of the Second Affiliated Hospital of Zhejiang University School of Medicine; and Huang Rufang, Founder and Director of the Cord Rare Disease Center, delivered speeches on behalf of the three organizing parties. Zhang Shuyang, President and Deputy Party Secretary of Peking Union Medical College Hospital; Xiang Yu, CEO of Langyu Group; and Harvey F. Lodish, Member of the U.S. National Academy of Sciences and Professor of Biology and Biological Engineering at the Massachusetts Institute of Technology, addressed the conference as distinguished guests. Meng Dongping, Party Secretary and Vice President of the China Chamber of Commerce for Import and Export of Medicines and Health Products; Wu Zhiying, Vice President of the Second Affiliated Hospital of Zhejiang University School of Medicine; and Huang Rufang, Founder and Director of the Cord Rare Disease Center, each delivered keynote presentations. Experts from previous editions also shared their reflections and well-wishes via video messages.
The conference will feature separate exchanges and discussions on various themes, including rare disease policies, gene therapy for rare diseases, and clinical translation in rare diseases. Below are selected excerpts from the forum on “Gene Therapy for Rare Diseases: Applications and Prospects”:

Bi Changhao, Group Leader of the Synthetic Biology Technology Research Group, Tianjin Institute of Industrial Biotechnology, Chinese Academy of Sciences
As is well known, gene editing is currently a highly prominent field, with two scientists being awarded the 2020 Nobel Prize in Chemistry for their contributions. Why has gene editing garnered such significant attention? Because it represents a highly suitable therapeutic approach for genetic diseases. Currently, gene therapy strategies are categorized into two types: ex vivo therapy, which involves editing cells outside the body and then reinfusing them into the patient for treatment; and in vivo therapy, which, similar to conventional large-molecule and small-molecule drugs, delivers therapeutic agents directly into the human body via delivery systems for treatment.
Currently, the more widely used second-generation gene editing technology is the base editor developed by David Liu.The chemical nature of base editors is a protein complex. Its function is to position the complex at specific locations on DNA to catalyze deamination reactions, thereby converting A-T base pairs into G-C base pairs. Compared with first-generation gene therapies, the advantages of second-generation base editing lie in its high efficiency of single-site mutations; most target sites achieve editing efficiencies exceeding 90%. Furthermore, its delivery is relatively simple, as it does not require a repair template.
Since the advent of base editors, the field of gene editing has been significantly advanced, prompting a surge in related research.Optimize each of its components or the entire assembly to create a broader range of novel base editors or gene-editing tools with additional functionalities.We have also developed a third-generation gene base editor on this basis, which enables the conversion of GC base pairs to CG base pairs. Its improvement lies in editing only the C6 position. Previous base editors had a certain editing window, which could bring some by-products while accurately correcting mutations, causing adverse effects.
Following the emergence of the third-generation editor, previous studies have revealed that this editor exhibits a strong sequence dependence. To address this issue, weIntroduction of High-Throughput Machine Learning Methods: First, a cell library comprising approximately 10,000 edited sites is constructed. These sites within the library of 10,000 cells are then subjected to high-throughput editing, and the entire edited library is collected. Subsequently, deep sequencing is employed to rapidly acquire a large volume of gene-editing sequences and corresponding data. Deep learning and analysis of this data using neural networks enable the generation of weight maps. However, as specificity and efficiency still require optimization, we have developed a next-generation editor based on this platform, which demonstrates further improvements in both efficiency and specificity.
Because base editors enable the correction of single-site mutations, they are currently the most important biological tool for rare diseases, particularly genetic disorders.For example, in the treatment of Leber congenital amaurosis, base editing offers a theoretically straightforward approach: correcting an adenine (A) to a guanine (G) using an ABE editor can achieve therapeutic efficacy. However, if there are other adenine bases near the target site, current base editors will also edit these nearby adenines, resulting in what is known as byproducts.
The next technology is single-window base editors, which can edit only the desired target sites, thereby addressing the issue of byproducts.This editor is currently in the research and development phase, and it still has a long way to go before entering clinical trials and obtaining subsequent regulatory approval for market launch. At present, through exchanges with the Cord Rare Disease Center, we have obtained certain information and focused on developing several specific sites. For progeria, we targeted 10 sites, achieving a correction rate of 40%–50%, which theoretically offers a complete cure for the disease. Although efficacy is expected to decline somewhat when transitioning from animal studies to human applications, current research results indicate that this decline will not be significant, and the data for these sites are promising. In the future, we will continue to optimize base editors to provide more precise and efficient editing tools.
There are approximately 7,000 rare diseases worldwide.
More than 80% of rare diseases are genetic disorders with known single-gene mutations.

Jiang Ruhong, Founder, Chairman, President, and Chief Executive Officer of ASCTx
Why Are Gene Therapies for Rare Diseases More Promising? There are over 7,000 rare diseases worldwide, and more than 80% of them are genetic disorders caused by known single-gene mutations. Therefore, they can potentially be targeted using gene therapy approaches.
However, in reality, very few related drugs have been approved; only 5%–6% of drugs are targeted at rare diseases.Moreover, the majority are small-molecule drugs. Traditional small-molecule drugs typically function by alleviating patients’ symptoms rather than curing the disease, whereas gene therapy has the potential to correct underlying genetic defects, thereby achieving a curative effect. Furthermore, successful gene therapy may require only a single dose to achieve lifelong disease improvement, eliminating the need for continuous treatment.
Currently, only two in vivo gene therapies have been marketed, and the field of gene therapy is still in its early stages of development. Developing a gene therapy drug requires careful consideration of the following aspects: First, regardless of the type of drug, a delivery system is essential; therefore, an effective and specific gene delivery system is a prerequisite, which currently falls into two main categories: viral and non-viral delivery systems. Second, tissue-specific promoters are required, including those for the liver, nervous system, skeletal muscle, eyes, and other tissues. Third, optimization systems for the target gene must be addressed, covering nucleotide sequence codon optimization, RNA transfection, protein synthesis, and protein secretion. Fourth, manufacturing processes for serotypes need to be developed; our company focuses on in vivo gene therapy, primarily on the development of manufacturing processes for adeno-associated virus (AAV) gene therapy products. Finally, analytical methods, regulatory requirements, and other related issues must also be comprehensively considered.
In terms of laws and regulations, the FDA’s primary regulations on gene therapy are mainly reflected in three aspects.
1. Regulations on Product Development, during product development, it is necessary to establish analytical methods for characterizing product-related variants and impurities; to establish evaluation criteria for multiple product attributes related to potency testing during initial clinical studies; to define manufacturing standards required for commercial-scale production; and to demonstrate product usability prior to the initiation of clinical trials.
2. Regulations on Preclinical Studies, during the research and development process, it is necessary to determine the range of biologically active doses, assess the feasibility, rationality, and safety of the clinical route of administration, and establish dosing regimens such as the initial clinical dose and dose escalation plan.
# Final Notes on Product Research, including the study population, trial design, dose selection, efficacy endpoints, and experience data.
In addition to the extensive regulatory and legal provisions, an even greater challenge lies in the fact that many technologies within the gene therapy field are novel. Yet, as a rapidly evolving sector, it inevitably gives rise to discrepancies between reality and expectations.In particular, the establishment of Chemistry, Manufacturing, and Controls (CMC) for gene therapy presents certain challenges. It may face a lengthy and complex manufacturing process, relatively low batch yields that are difficult to scale up, cumbersome release testing, and prolonged production timelines. From a clinical perspective, patient selection for enrollment also poses a significant challenge. As gene therapy products enter the cell nucleus, current safety considerations necessitate the establishment of a systematic approach to properly select patients prior to enrollment. The FDA requires a five-year follow-up period to confirm safety and efficacy.
Currently, ASC-618, a second-generation gene therapy for hemophilia A, received FDA clinical trial approval on July 7 this year. It treats hemophilia A by delivering the target gene in vivo via an adeno-associated virus (AAV) vector. ASC-618 utilizes a bioengineered factor VIII to achieve efficient biosynthesis and secretion, ultimately elevating factor VIII levels in the blood at a lower effective dose to attain therapeutic efficacy. Furthermore, the potential for low-dose administration of ASC-618 significantly reduces production costs and markedly improves the sustainability of therapeutic effects.
Finally, since our inception, we have progressed step by step—from building and applying our technology platform to now preparing for clinical enrollment. Throughout this journey, our goal has been to deliver compliant and effective gene therapies to patients with rare diseases as rapidly as possible.
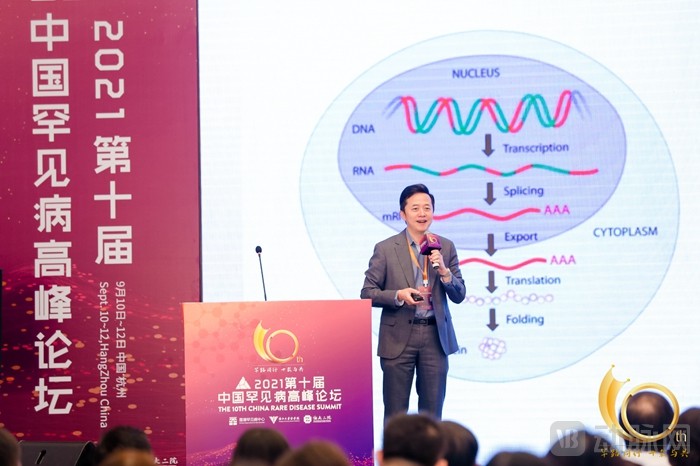
Wei Dong, CEO of Ediacare
Since the discovery of the DNA double helix structure by scientists in 1953, humanity has been contemplating whether diseases could be treated at the genetic level. Particularly now, with gene sequencing becoming more accessible and affordable, our understanding of disease mechanisms has become significantly clearer.Genomic drugs primarily include gene therapy, genetically engineered cell therapy, and gene editing. Unlike traditional small-molecule and large-molecule drugs, which mainly act at the protein level, this class of therapeutics operates at the DNA and RNA levels, thereby addressing diseases at their root cause.
In China, gene editing was still a rather mysterious-sounding concept just a few years ago. Editing genes is akin to modifying a few letters within a massive dictionary containing three billion characters. How, then, can this be achieved?This involves two aspects: first, knowing the editing sites, which ensures precision; second, having editing tools to perform the edits.Why was gene editing previously considered so challenging? Because earlier gene-editing tools essentially performed the two aforementioned tasks simultaneously, resulting in considerable technical difficulty. Later, scientists decoupled these two processes and developed CRISPR technology, which spurred rapid advancement across the entire field of gene editing. The pioneers of this technology were awarded the Nobel Prize in Chemistry in 2020.
When it comes to therapies, the primary concerns are always safety and efficacy, and gene-editing therapies are no exception. First, regarding safety: from 2019 to the present, we have reviewed at least six publications related to gene editing and have not identified any significant safety risks associated with gene editing. In terms of efficacy, data presented by Vertex Pharmaceuticals and CRISPR Therapeutics in June this year at the European Hematology Association (EHA) Congress showed that among all 15 patients treated with a CRISPR-based ex vivo gene-editing therapy for transfusion-dependent β-thalassemia, none required further blood transfusions after treatment. Additionally, among all 7 patients treated for sickle cell disease, none experienced vaso-occlusive crises after treatment.
Gene editing technology has also demonstrated future potential in the preliminary efficacy of universal CAR-T therapies. In the realm of in vivo gene editing, Intellia Therapeutics and Regeneron released treatment data for patients with transthyretin amyloidosis (ATTR) this June, showing a significant reduction in levels of the pathogenic protein and highlighting the potential for a one-time curative treatment. Looking ahead, we can expect to see broader applications of gene editing technology.
So, how is gene editing technology developing in China? Currently, there are more than 10 publicly listed gene editing companies in the United States, and new gene editing startups are securing financing rounds of $100 million or more. In contrast, China’s industry is still in its early stages of development.
Since 2018, funding in China’s gene-editing sector has remained relatively subdued in recent years due to a controversial ethical incident involving gene editing.It can be said that China’s basic scientific research capabilities in gene editing are on par with the world’s most advanced levels; however, product translation has remained at the stage of investigator-initiated clinical trials.Certainly, as regulatory frameworks strengthen and improve, the investment environment and translational capabilities for gene editing in China are gradually improving. In January this year, Boya Jiayin’s first gene-editing product, ET-01, a research therapy targeting transfusion-dependent β-thalassemia, received approval from the National Medical Products Administration (NMPA) for its Investigational New Drug (IND) application. This is currently the only approved IND for a gene-editing therapy in China. Last week, we also announced the enrollment of the first patient.
In 2018, we began establishing our company’s CMC, preclinical research, clinical research, and regulatory affairs teams. After two years of relentless effort, we successfully filed the Investigational New Drug (IND) application for China’s first gene-editing therapy. Meanwhile, the Institute of Hematology & Blood Diseases Hospital, Chinese Academy of Medical Sciences (also known as the Institute of Hematology, Chinese Academy of Medical Sciences), conducted investigator-initiated clinical trials using our product. The Institute has reported on these trials; in May this year, the first patient was discharged and achieved transfusion independence.
Furthermore, from the perspective of gene editing technology, we have established a strategic presence in both DNA editing and RNA editing. In the field of RNA editing, we are utilizing the LEAPER technology developed by the laboratory of Professor Wei Wensheng at Peking University, who is the scientific founder of our company.We have successfully established this system using vectors such as AAV and LNP, achieving robust gene editing efficiency. We aim to advance RNA base editing therapies along the trajectory of gene therapy to benefit a broader patient population. Our goal is for gene editing to provide a unique approach that directly corrects disease-causing mutations in patients’ own genes. Furthermore, we aspire to leverage our therapeutic products to assist more individuals with genetic disorders and those at high risk.