China Pharmaceutical University's Xu Yungen/Zou Yi Team Reports Spirocyclic Quinoline-Based Oral Small-Molecule TNF-α Inhibitor XS-18 with Clinical Potential for Rheumatoid Arthritis

AbbVie
Innovative Drug Developer
PharmaView
2026.03.31 Online


Recently, Xu Yungen/Zou Yi Team from China Pharmaceutical UniversityPublished an article titled in the authoritative journal of medicinal chemistry, JMC"Discovery of Quinoline-based Oral Small-molecule Tumor Necrosis Factor α (TNF-α) Inhibitors with a Spirocyclic Scaffold"Research Paper.
This study addresses the clinical challenges associated with biologics in rheumatoid arthritis (RA) treatment, such as high infection risk, increased tumor incidence, and inconvenient administration. Based on a scaffold-hopping strategy, structural optimization was performed on AbbVie-CPD-12 to design and synthesize a series of small-molecule TNF-α inhibitors containing quinoline scaffolds and spirocyclic structures. Among these, the candidate compound XS-18 demonstrated excellent TNF-α binding affinity (FP IC₅₀ = 123 nM; Kᴅ = 45.9 nM) and significantly inhibited TNF-α-mediated inflammatory pathways in vitro. In a collagen-induced arthritis (CIA) mouse model, orally administered XS-18 exhibited potent anti-inflammatory effects, with efficacy in promoting articular cartilage repair surpassing that of the clinical drug tofacitinib. Importantly, XS-18 displayed favorable pharmacokinetic properties (T₁/₂ = 7.1 h, oral bioavailability F = 56.8%) and good in vivo safety, providing a promising new candidate molecule with clinical translational potential for the development of orally active small-molecule TNF-α inhibitors to treat autoimmune diseases.
1min Quick View
Research Background
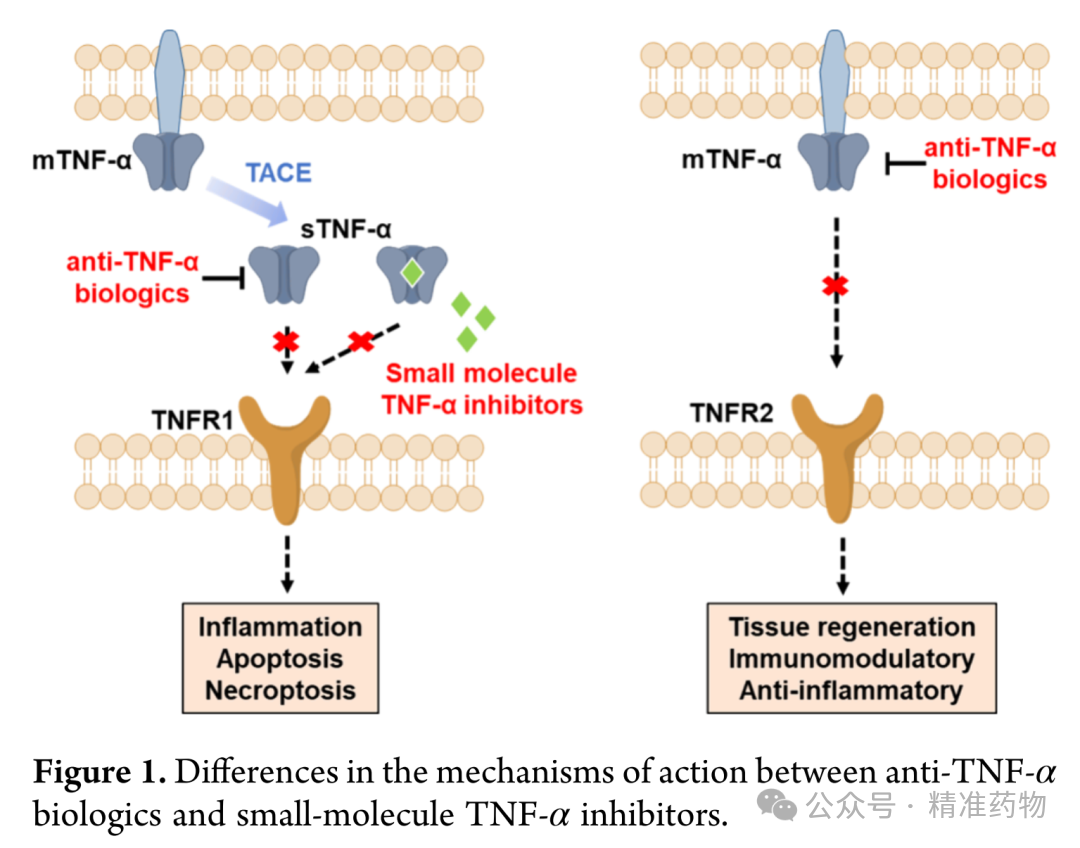
Rheumatoid arthritis (RA) is a chronic progressive autoimmune disease, with early symptoms of joint swelling and pain, and later leading to cartilage destruction and bone erosion, ultimately causing joint deformities and functional impairment, severely affecting the quality of life of patients. Tumor necrosis factor α (TNF-α) is an important target for RA treatment. Currently used anti-TNF-α biologics in clinical practice (such as infliximab, etanercept, and adalimumab) have proven efficacy but are limited by increased risks of serious infections, elevated tumor incidence, and the need for injectable administration. Small-molecule TNF-α inhibitors offer advantages such as convenient oral administration, lower production costs, and reduced risk of severe side effects, and can selectively target the soluble TNF-α/TNFR1 signaling pathway without affecting TNFR2-mediated tissue repair functions. Therefore, the development of novel oral small-molecule TNF-α inhibitors holds significant clinical value. However, only a few small-molecule TNF-α inhibitors have entered clinical trials, and no such drugs have been approved for marketing yet, indicating that clinical needs remain largely unmet.
Key Content
The research team conducted rational drug design based on AbbVie-CPD-12 using a scaffold hopping strategy. By combining the quinoline scaffold of BMS-CPD-42 with an optimized spiro structure through molecular docking analysis, they obtained the candidate compound XS-18 after multiple rounds of structural optimization. This compound exhibited excellent TNF-α binding affinity (FP IC₅₀ = 123 nM, Kᴅ = 45.9 nM) and TNF-α/TNFR1 interaction inhibitory activity (IC₅₀ = 36.4 nM). In vitro, XS-18 effectively inhibited L929 cell apoptosis, blocked the NF-κB signaling pathway, and suppressed the expression of the inflammatory cytokine IL-6, with anti-inflammatory activity superior to that of AbbVie-CPD-12. It also demonstrated favorable pharmacokinetic properties (oral bioavailability of 56.8%, half-life of 7.1 hours) and in vivo safety. In a CIA mouse model, oral administration of XS-18 significantly reduced joint swelling and arthritis scores. Its cartilage repair effects were better than those of the clinical drug tofacitinib, and it effectively downregulated the expression of IL-6 and TNF-α in ankle joint tissues, showing promising potential for clinical translation as an orally administered small-molecule TNF-α inhibitor for the treatment of rheumatoid arthritis.
Research Summary
This study successfully designed a novel quinoline-based spiro small molecule TNF-α inhibitor, XS-18, through a scaffold-hopping strategy. The compound has the following notable characteristics: (1) Excellent target binding affinity, capable of directly binding to TNF-α and disrupting its trimer symmetry, selectively inhibiting the sTNF-α/TNFR1 interaction; (2) Significant anti-inflammatory activity both in vitro and in vivo, showing superior anti-inflammatory effects compared to the reference compound at the cellular level and in the CIA animal model, particularly excelling in cartilage repair over the clinical drug tofacitinib; (3) Favorable pharmacokinetic properties, with an oral bioavailability of 56.8% and a half-life of 7.1 hours, supporting oral administration; (4) Good safety profile, with no obvious toxic reactions observed in acute and subacute toxicity studies. XS-18, as an orally administrable small molecule TNF-α inhibitor with clinical translational potential, provides a new candidate drug for the treatment of autoimmune diseases such as rheumatoid arthritis, with the potential to overcome the limitations of existing biologics and offer patients a safer and more effective treatment option. Additionally, this research provides a successful example for developing protein-protein interaction inhibitors based on the scaffold-hopping strategy.
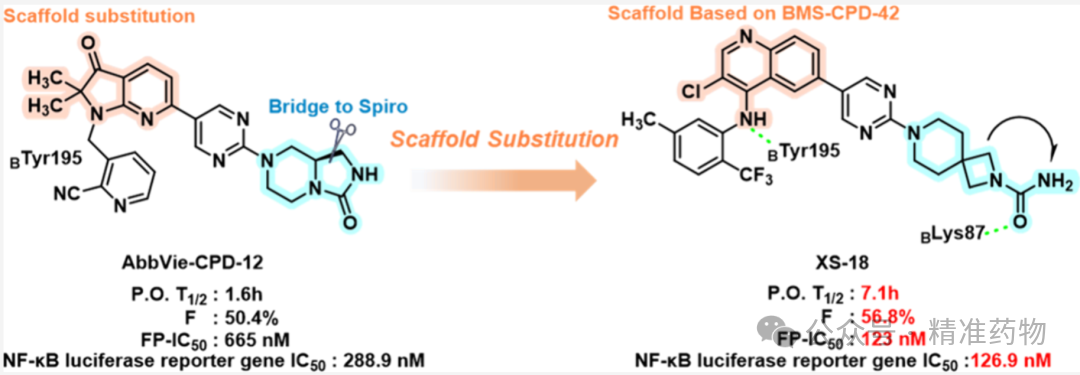
Image Source: ACS
Read in detail
01
INTRODUCTION
Research Background
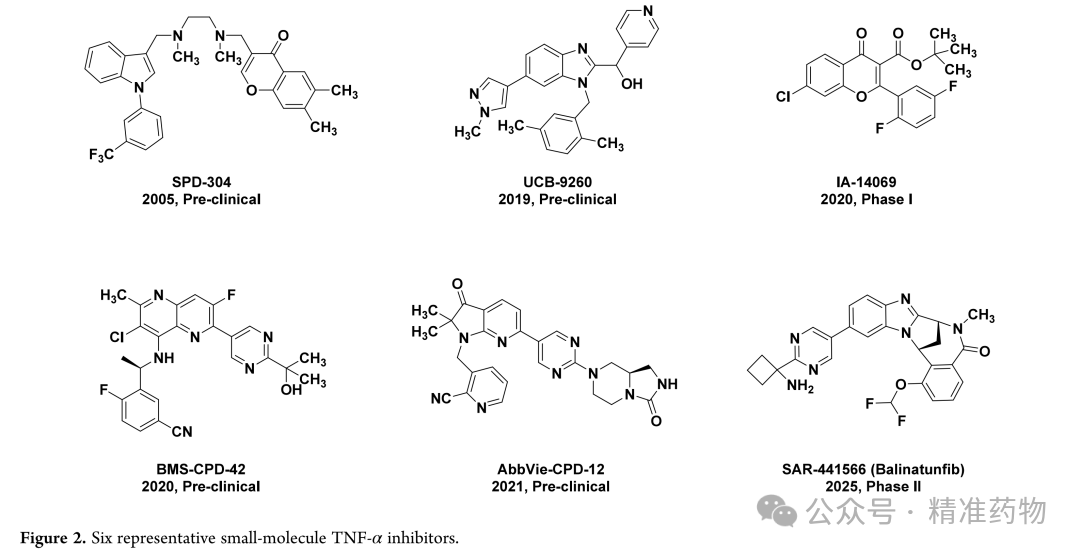
Rheumatoid arthritis is a chronic progressive autoimmune disease, with early manifestations of joint swelling and pain, and later leading to cartilage destruction and bone erosion, ultimately causing joint deformity and functional impairment. Tumor necrosis factor α (TNF-α) in rheumatoid arthritis patients is mainly secreted by M1 macrophages and mediates the inflammatory process. Targeting TNF-α is a mature and effective strategy for treating this type of disease. Currently, several anti-TNF-α biologics have been approved for clinical use, but they carry adverse effects such as an increased risk of severe infections and tumors. TNF-α can be divided into two forms: transmembrane and secreted, which bind to TNFR1 and TNFR2 receptors, respectively. Anti-TNF-α biologics lack selectivity for these two forms, simultaneously inhibiting both TNFR1 and TNFR2 pathways, while the TNFR2 pathway mediates tissue repair. This non-selective action leads to adverse reactions. In contrast, small-molecule TNF-α inhibitors selectively target the secreted TNF-α/TNFR1 pathway, offering advantages such as oral convenience, low cost, and minimal side effects. To date, six representative small-molecule inhibitors have been discovered, with only two entering clinical trials, and no drugs have yet been approved. The development of novel small-molecule TNF-α inhibitors is of great significance. Based on AbbVie-CPD-12, this study designed and synthesized quinoline derivatives through scaffold replacement. XS-18 was identified as the most effective candidate compound, demonstrating significant anti-inflammatory efficacy in the CIA model and showing promise as an oral treatment candidate for rheumatoid arthritis.
02
RESULTS AND DISCUSSION
① Rational Design Based on Skeleton Replacement
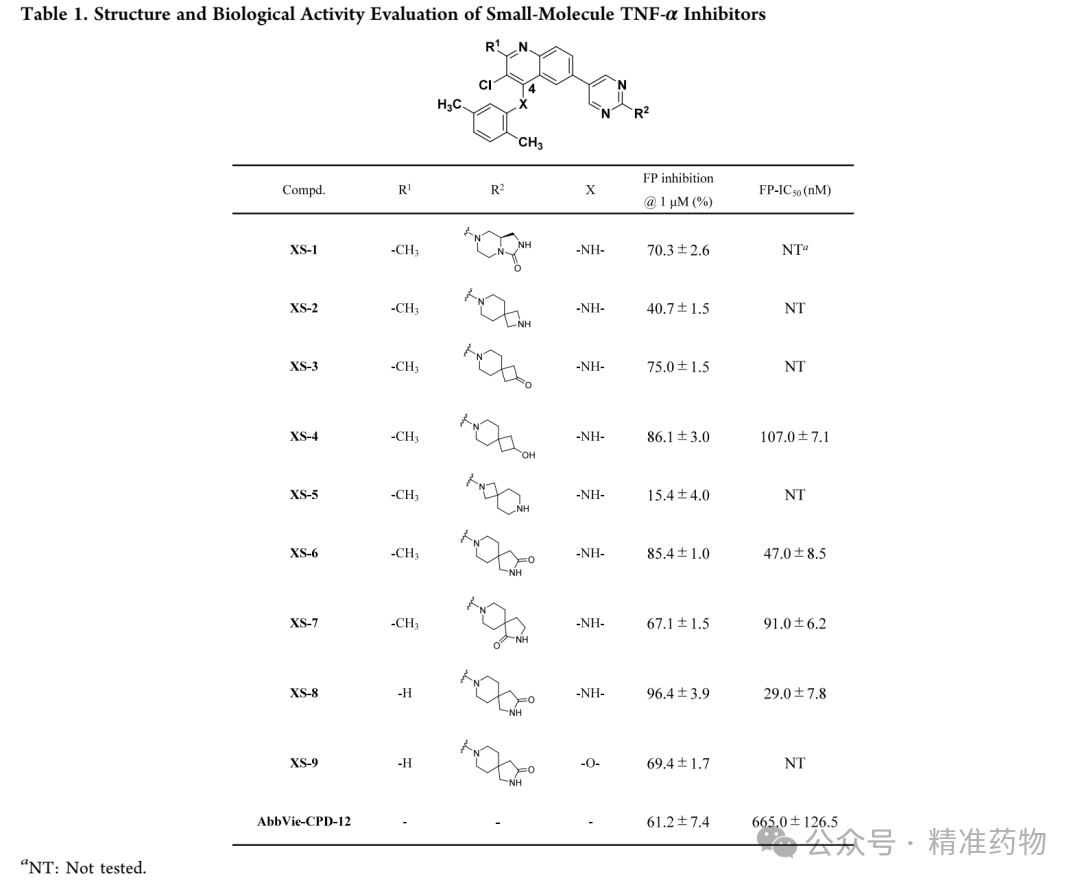
The research team first analyzed the predicted binding modes of AbbVie-CPD-12 and BMS-CPD-42 with the TNF-α protein (PDB code: 7JRA) to investigate the binding patterns of their different structural scaffolds. The results showed that, within the hydrophobic pocket of TNF-α, the 1,2-dihydro-3H-pyrrolo[2,3-b]pyridin-3-one scaffold of AbbVie-CPD-12 forms hydrogen bonding interactions with CTyr227 and π-π stacking interactions with CTyr135. In contrast, the 1,5-naphthyridin-4-amine scaffold of BMS-CPD-42, in addition to the aforementioned interactions, also forms an additional hydrogen bond through its -NH group with BTyr195 and π-π stacking interactions between the aniline group and ATyr135. Therefore, compared with AbbVie-CPD-12, BMS-CPD-42 participates in more significant molecular interactions within the TNF-α binding pocket, while AbbVie-CPD-12 primarily engages in single hydrophobic interactions. In the solvent-accessible channel, AbbVie-CPD-12 better occupies this region by introducing a hydrophilic bridged-ring structure compared to the tertiary alcohol group of BMS-CPD-42; however, the bridged ring fails to form critical interactions with adjacent polar amino acid residues such as BLys87. To address this limitation, the research team replaced the scaffold of AbbVie-CPD-12 with that of BMS-CPD-42 to enhance the interactions of the small molecule within the TNF-α binding pocket, while optimizing the bridged-ring structure located in the solvent-accessible channel to promote critical interactions with adjacent polar amino acid residues.
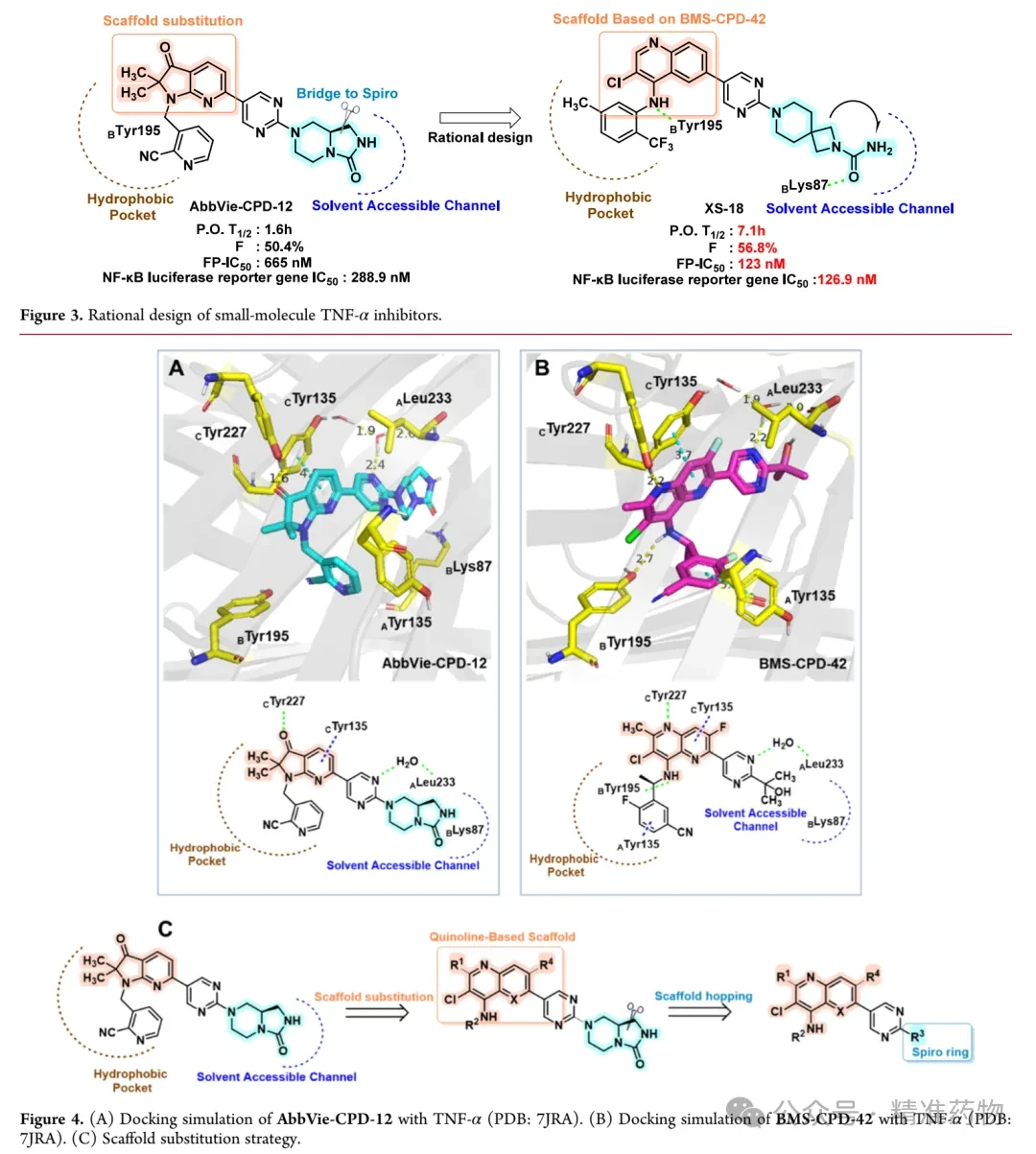
② Structure-Based Optimization
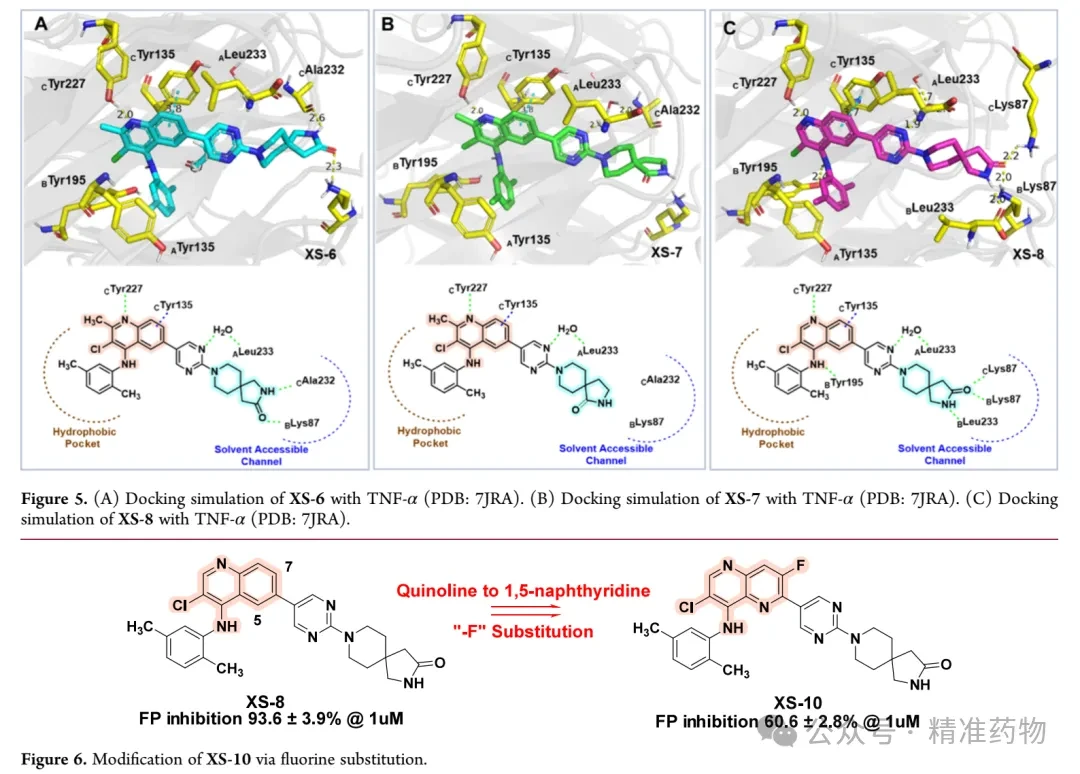
This study used fluorescence polarization (FP) binding experiments to screen the binding affinity of small molecules with TNF-α. To address the high false-positive rate issue of the FP probe used in AbbVie-CPD-12, the research team redesigned a high-affinity FP probe using quinoline as the core scaffold. First, they replaced the pyrrolopyridone core of AbbVie-CPD-12 with 2-methylquinoline and modified the bridged-ring system by introducing a spiro structure with polar functional groups. XS-1 showed enhanced activity after the introduction of a 2,5-dimethylaniline group at the 4-position, confirming the feasibility of the strategy. XS-2 adopted a 2,7-diazaspiro[3.5]nonane occupying the solvent channel but exhibited weaker activity. Replacing "-NH-" with a carbonyl or "-CH(OH)-" yielded XS-3 and XS-4, which demonstrated significantly increased affinity, indicating that hydrogen bond donors are more beneficial for binding. XS-5, which coupled azetidine with a pyrimidine ring, showed reduced activity due to increased steric hindrance. XS-6 and XS-7, which introduced spiroamide groups, outperformed AbbVie-CPD-12, but XS-7 showed decreased activity due to the loss of hydrogen bonding interactions with CAla232 and BLys87. XS-8, with the R1 position’s "-CH3" replaced by "-H", exhibited further enhanced binding affinity (FP IC₅₀ 29 nM), and molecular docking revealed that it eliminated steric hindrance, formed a hydrogen bond with BTyr195, and established more interactions within the solvent channel. XS-9, where "-NH-" was replaced with "-O-", showed reduced affinity, confirming the importance of this hydrogen bond. XS-10, which replaced quinoline with 1,5-naphthyridine and introduced fluorine atoms, did not improve affinity and instead led to decreased activity.
③ Optimization of In Vitro Metabolic Stability
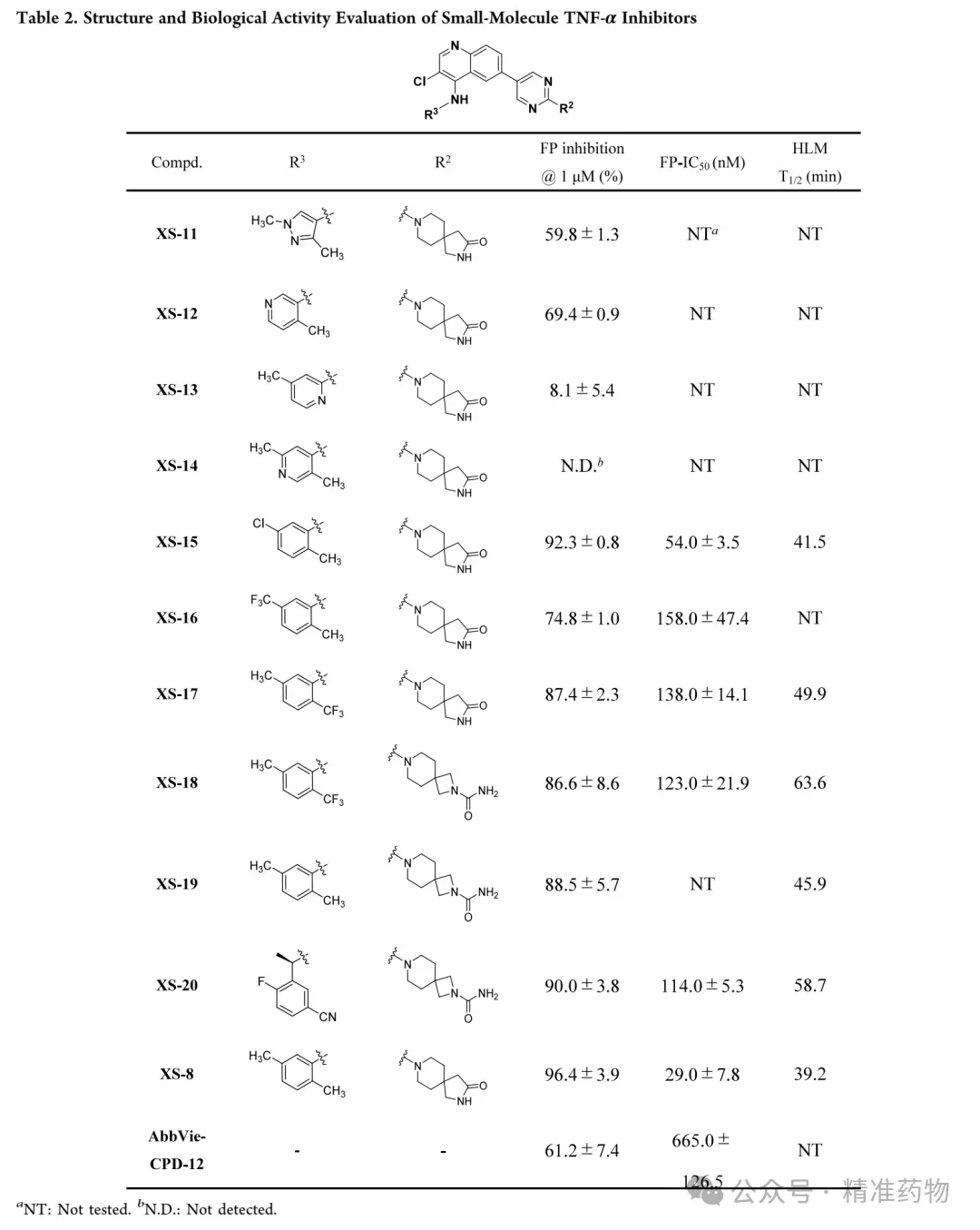
In vitro human liver microsomal stability assays showed that XS-8 had a short half-life (T₁/₂ = 39.2 min). The research team optimized it by introducing electron-withdrawing groups onto the 2,5-dimethylaniline scaffold. XS-11, which replaced the structure with a pyrazole ring, showed significantly reduced activity; XS-12 and XS-13, where methyl groups were removed, exhibited decreased affinity, confirming the importance of the methyl group at the 2-position. XS-14, with a nitrogen atom directly introduced, almost completely lost activity. XS-15, incorporating a chlorine atom, retained excellent activity (FP IC₅₀ = 54 nM), while XS-16 and XS-17, featuring trifluoromethyl groups, demonstrated moderate activity with half-lives extended to 41.5 min and 49.9 min, respectively. Considering the improvement in oral bioavailability due to the trifluoromethyl group, XS-17 was chosen for further optimization. Replacing the five-membered spirocycle with a four-membered ring and substituting the amide with a urea group resulted in XS-18, which displayed significantly enhanced metabolic stability (T₁/₂ = 63.6 min) while maintaining inhibitory activity. XS-19 confirmed that urea modification could improve metabolic stability but slightly reduced activity, while XS-20, modified based on BMS-CPD-42, showed increased activity but decreased metabolic stability. Given the favorable metabolic profile and inhibitory activity of XS-18, it was selected for subsequent in vitro anti-inflammatory activity evaluation.
④XS-18 Significantly Inhibits TNF-α-Mediated Inflammatory Pathway In Vitro
To evaluate the differences in in vitro anti-inflammatory activity between XS-18 and AbbVie-CPD-12, the research team conducted L929 cell apoptosis neutralization assays and HEK293 cell NF-κB luciferase reporter gene assays. In the presence of TNF-α (10 ng/mL) and actinomycin D, L929 cells undergo apoptosis via the TNF-α/TNFR1/caspase-8 signaling pathway, and disrupting the interaction between TNF-α and TNFR1 significantly reduces the apoptosis rate. At a concentration of 1 μM, XS-18 inhibited L929 cell apoptosis by 68.1±9.8%, compared to 60.8±10.8% for AbbVie-CPD-12 under the same conditions. Moreover, XS-18 maintained nearly 50% inhibition at a lower concentration of 1.37 nM, indicating its superior potency over AbbVie-CPD-12. Activation of the TNF-α/TNFR1 signaling pathway leads to NF-κB nuclear translocation, promoting the transcription of downstream inflammatory factors and exacerbating inflammatory responses. Therefore, the in vitro anti-inflammatory activity of small-molecule TNF-α inhibitors can be assessed by detecting the inhibitory effect on NF-κB-dependent luciferase activity in HEK293 cells stably transfected with an NF-κB reporter gene. First, the cytotoxicity of XS-18 on NF-κB-TA-Luc HEK293 cells was evaluated, showing that cell viability remained above 95% after 72 hours of incubation at 20 μM, indicating minimal toxicity and good biocompatibility of XS-18 for this cell line. The luciferase assay demonstrated that XS-18 effectively inhibited NF-κB reporter gene expression, with an IC₅₀ value of 126.9±33.8 nM, significantly lower than that of AbbVie-CPD-12 (IC₅₀: 288.9±140.0 nM).
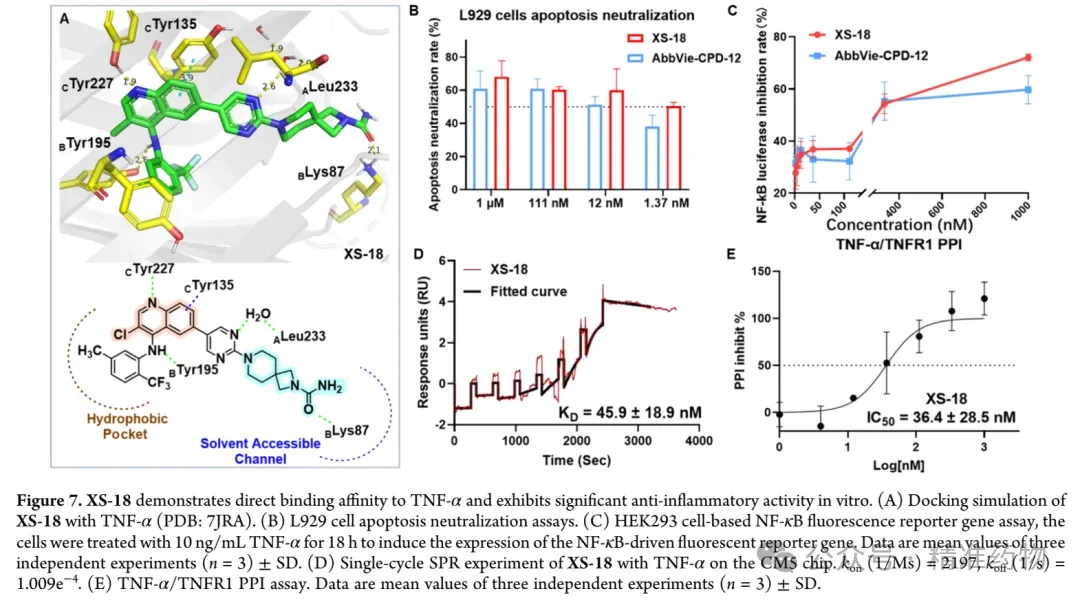
Image Source: ACS
⑤XS-18 inhibits IκBα phosphorylation and IL-6 expression in human rheumatoid arthritis fibroblast-like synoviocytes MH7A
IκBα is a key regulatory protein in the TNF-α/TNFR1/NF-κB signaling pathway. When this pathway is not activated by TNF-α, IκBα can inhibit NF-κB function and prevent its nuclear translocation. After the pathway is activated by TNF-α, IκBα undergoes phosphorylation, ubiquitination, and subsequent degradation, leading to the release of NF-κB, which enters the nucleus to initiate the transcription of target genes such as IL-6, IL-1β, and TNF-α, promoting the occurrence and development of inflammation. Fibroblast-like synoviocytes in rheumatoid arthritis, as major participants in joint inflammation, can promote the progression of inflammation through the aforementioned signaling pathway, resulting in cartilage tissue damage and joint dysfunction. Based on this, the research team selected human rheumatoid arthritis fibroblast-like synoviocytes MH7A and used Western Blot to evaluate the inhibitory effect of XS-18 on IκBα phosphorylation in the TNF-α/TNFR1/NF-κB signaling pathway. The results showed that stimulation of MH7A cells with TNF-α (10 ng/mL) for 15 minutes significantly increased the p-IκBα/IκBα ratio, while XS-18 at concentrations of 5 μM and 10 μM significantly decreased the p-IκBα/IκBα ratio. Moreover, at 5 μM, XS-18 exhibited a more pronounced inhibitory effect on p-IκBα/IκBα than AbbVie-CPD-12 at the same concentration. This result more directly demonstrates that XS-18 can block TNF-α-mediated activation of the TNFR1/NF-κB inflammatory pathway by preventing IκBα phosphorylation, thereby blocking NF-κB nuclear entry and transcription of related pro-inflammatory target genes. Real-time quantitative PCR results further indicated that XS-18 downregulated IL-6 expression as expected. MH7A cells inherently express IL-6, and after stimulation with TNF-α (10 ng/mL) for 15 minutes, IL-6 expression was upregulated. Both 5 μM and 10 μM concentrations of XS-18 significantly inhibited IL-6 expression, with effects superior to those of AbbVie-CPD-12, indicating that XS-18 possesses superior anti-inflammatory properties.
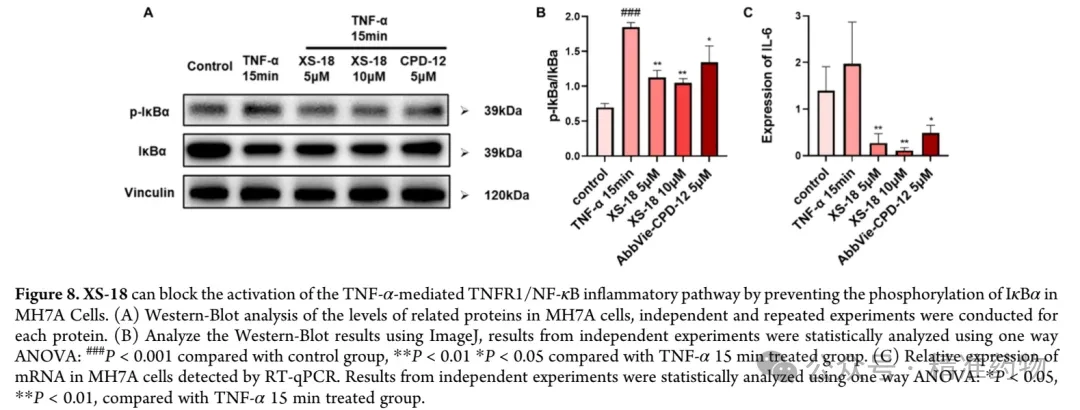
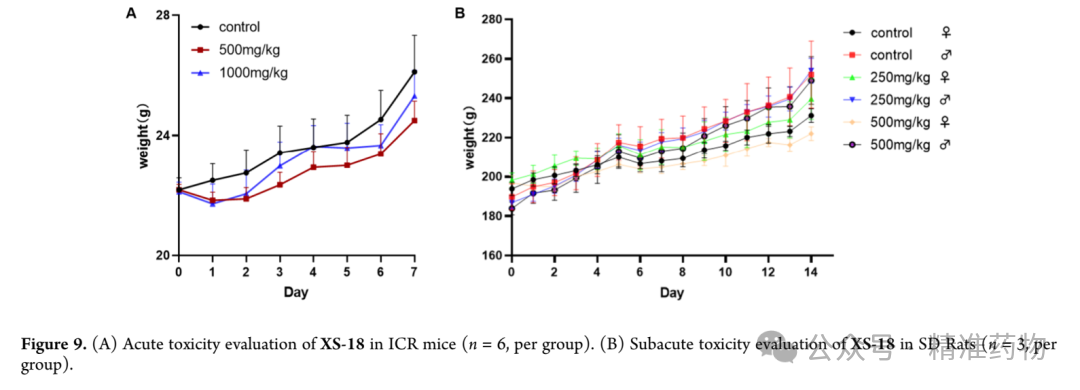
⑥In vivo pharmacokinetics and safety evaluation of XS-18
The research team evaluated the pharmacokinetic properties of XS-18 in rats. The results showed that XS-18 has excellent oral bioavailability (F = 56.8%) and a long half-life (T₁/₂ = 7.1 h), with good AUC₀₋∞ performance, suggesting its potential as an oral small-molecule TNF-α inhibitor. Based on the pharmacokinetic data, the research team further assessed the in vivo safety of XS-18 through acute toxicity tests in mice and subacute toxicity tests in rats: In the ICR mouse acute toxicity test, no significant weight loss or abnormal behavioral changes were observed during the 7-day observation period after a single oral administration of 500 mg/kg or 1000 mg/kg; in the SD rat subacute toxicity test, after 14 consecutive days of daily oral administration of 250 mg/kg and 500 mg/kg, there was no significant weight loss in the treatment groups compared to the control group, and analysis of major organ indices such as heart, liver, spleen, lung, and kidney showed no statistically significant differences in organ-to-body weight ratios between groups, indicating that XS-18 did not cause detectable organ toxicity under the experimental conditions.

⑦XS-18 In Vivo Anti-Inflammatory Efficacy Evaluation
The research team evaluated the in vivo efficacy of XS-18 using a collagen-induced arthritis mouse model (CIA) and compared it with tofacitinib, the first approved oral targeted synthetic disease-modifying antirheumatic drug (tsDMARD) for the treatment of rheumatoid arthritis; tofacitinib, as a pan-JAK inhibitor, is associated with an increased risk of major cardiovascular events and malignancies. DBA/1J mice successfully established the rheumatoid arthritis model after two collagen stimulations. Based on the pharmacokinetic characteristics of XS-18, three dose levels of 5, 15, and 50 mg/kg were selected for twice-daily oral administration over 14 consecutive days, monitoring the average increase in hind paw volume. During the first 8 days (days 44-52), the hind paw swelling in the model group showed a decreasing trend, suggesting spontaneous healing activity, but from day 9 to day 14 (days 52-58), the rate of increase in hind paw volume began to rise, indicating that the self-healing capacity had reached a plateau. In the experiment, the medium-dose group (15 mg/kg) of XS-18 showed the most significant inhibitory effect on the increase in hind paw volume, comparable to the tofacitinib group after 14 days of treatment. Notably, the baseline level of hind paw volume increase at the start of treatment was lowest in the tofacitinib group. The high-dose group (50 mg/kg) of XS-18 also exhibited good inhibitory effects, but due to a higher initial baseline, the efficacy after 14 days was slightly lower than that of the medium-dose group. The low-dose (5 mg/kg) and medium-dose (15 mg/kg) groups had similar baseline levels, with the low dose showing relatively weaker inhibition of hind paw volume increase. In addition to the increase in hind paw volume, changes in claw thickness are also a key parameter for assessing the progression of rheumatoid arthritis. Since the baseline measurements at the start of treatment were similar across groups, the inhibitory effect of XS-18 on claw thickness showed a dose-dependent relationship; the high-dose group (50 mg/kg) significantly reduced claw thickness, comparable to the tofacitinib group, while the medium-dose group (15 mg/kg) also demonstrated statistically significant inhibitory effects. To comprehensively evaluate the occurrence and development of rheumatoid arthritis in mice, including clinical indicators such as ankle and paw redness and swelling, the joint conditions of mice in each drug treatment group were assessed to determine the arthritis score. The high-dose group (50 mg/kg) showed a significant reduction in arthritis score, superior to the tofacitinib treatment group, while the medium-dose group showed moderate reduction, and the low-dose group showed no obvious therapeutic effect. At the end of treatment, photographic records of the ankles and paws visually confirmed the significant anti-inflammatory effect of the high-dose XS-18 group, with marked relief of redness and swelling, surpassing the improvement seen in the tofacitinib group. No significant changes in body weight were observed in any group during the treatment period, and H&E staining of major organs revealed no obvious histopathological abnormalities, supporting the favorable safety profile of orally administered XS-18.
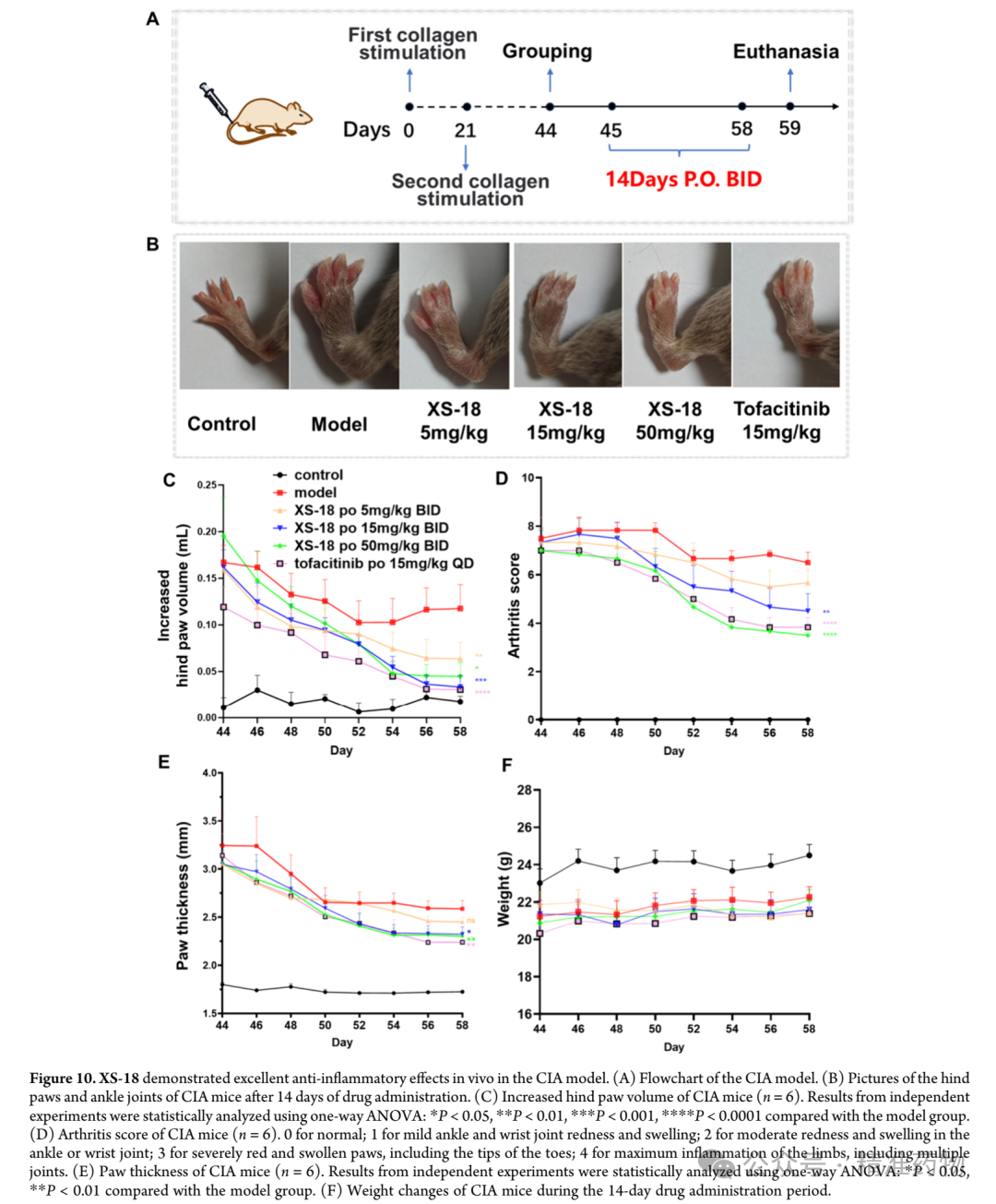
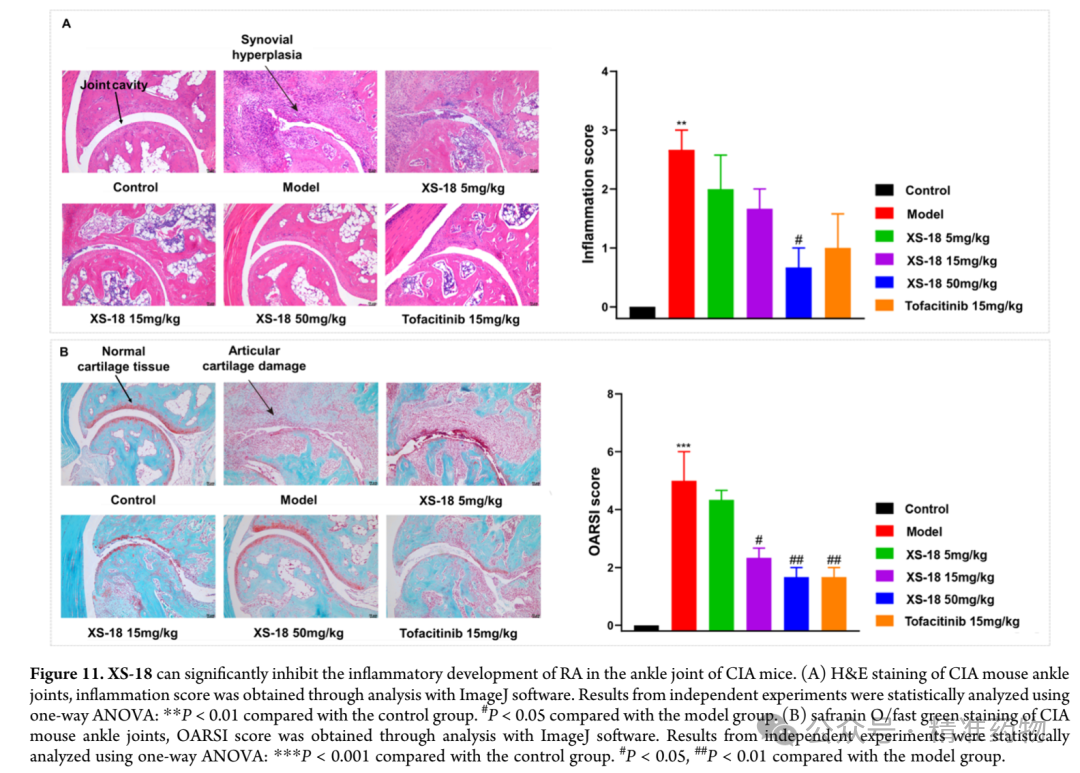
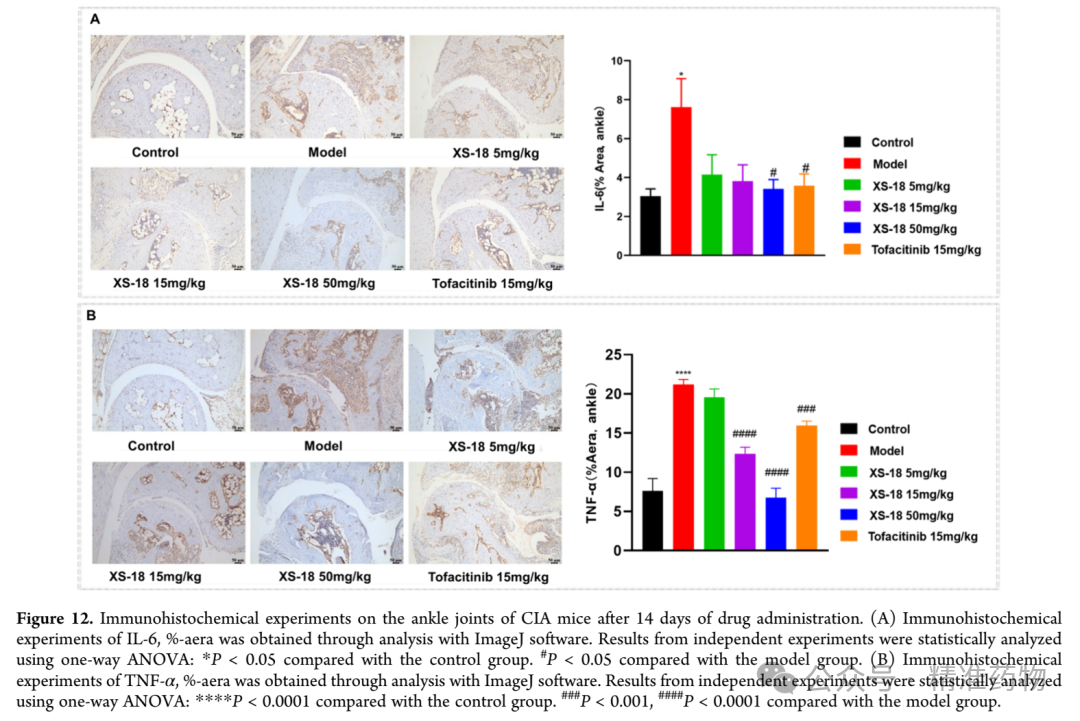
Image Source: ACS
Summary and Outlook
In summary, the research team successfully designed and synthesized XS-18 through a scaffold replacement strategy, a novel small-molecule TNF-α inhibitor derived from the previously reported AbbVie-CPD-12. XS-18 demonstrated strong binding affinity for TNF-α in FP and SPR assays and showed significant in vitro anti-inflammatory activity. Additionally, XS-18 exhibited favorable in vivo safety profiles and robust pharmacokinetic properties (T₁/₂ = 7.1 h, F = 56.8%), suggesting its potential as an oral TNF-α inhibitor. In the CIA model, XS-18 displayed remarkable anti-inflammatory activity by effectively suppressing the expression levels of IL-6 and TNF-α in ankle joint tissues, with therapeutic effects surpassing those of tofacitinib. These findings indicate that XS-18, as a small-molecule TNF-α inhibitor, holds promise as an effective oral tsDMARD, potentially offering an alternative to tofacitinib while reducing the risk of severe clinical adverse reactions. Given the complex pathogenic mechanisms underlying autoimmune diseases, other proteins such as TACE, receptor-interacting protein kinase 1/3 (RIP1/3), and IL-17A also play critical roles in inflammation development beyond TNF-α. Therefore, the development of dual-target inhibitors modulating TNF-α and other inflammation-related protein activities could provide more comprehensive control over autoimmune diseases.
For detailed and comprehensive information, please refer to the original article.:
https://doi.org/10.1021/acs.jmedchem.5c03457
Disclaimer: The publication/reposting of this article is solely for the purpose of information dissemination, and does not represent the views of this official account or confirm the authenticity of its content. Any judgment made based on this content will be at your own risk.If there is any infringement, it will be deleted upon notification!
Long press to follow this official account

