Tianrang AI's TRFold Ranks Second Globally in Protein Structure Prediction, Files for IPO
Major advancements in every stage of medical development are closely linked to breakthroughs in science and technology.
New drug development is one of the most risky, complex, and time-consuming areas of technological research in human progress. High R&D costs, long development cycles, and low success rates have long been the “three major burdens” weighing on pharmaceutical companies. According to a set of data published by the British journal Nature, the cost of developing a new drug is approximately $2.6 billion, the process takes about 10 years, and the success rate is less than one in ten.
The typical new drug development process usually includes the identification of drug targets and optimal compounds, preclinical studies, Phase I, II, and III clinical trials, and approval by the Food and Drug Administration. However, statistics show that the number of explorable molecules in the drug-like chemical space is as high as 1023Up to 1060, discovering new drugs can be likened to finding a needle in a haystack.
With the development of AI technology,AI applications across all stages of new drug development can significantly reduce trial-and-error and rework time while ensuring analytical quality, thereby enhancing R&D efficiency., paving the way for rapid and efficient new drug development while reducing R&D costs. Statistics show that AI-enabled drug discovery has reduced costs by 35% in some cases, shortening the development cycle from 5–10 years to 1–3 years.
Proteins are involved in executing nearly all cellular functions. To perform their specific roles, proteins must fold into precise three-dimensional structures. The three-dimensional structure of a protein directly determines its function; once this structure is disrupted, the protein loses its functionality. Many common diseases, such as cancer and Alzheimer’s disease, are caused by abnormalities in the structure of critical proteins within the body.
Protein structure prediction is a crucial branch of structural biology; however, existing experimental methods are insufficient to elucidate certain important protein structures, necessitating the use of additional bioinformatics and computational biology approaches for exploration.
AI applications in the field of protein structure have deciphered, through prediction, certain structures that could not be resolved by traditional observational methods. These predictions exhibit high confidence levels and closely approximate reality, which will significantly accelerate research in the life sciences. Furthermore, this advancement will drive improvements in healthcare, food sustainability, and emerging technologies, thereby further promoting progress in biological sciences, drug development, and synthetic biology.
This July, Google’sAI CompanyDeepMind Releases Source Code for Its AI System AlphaFold 2,and published a paper in NatureElaborates on the technical details of AlphaFold2, a system capable of accurately predicting the 3D structure of proteins based on amino acid sequences,It sent shockwaves through the biotech industry.
At CASP14 (the 14th Critical Assessment of Protein Structure Prediction) in 2020, AlphaFold2’s accurate prediction of protein structures was recognized as one of the Top Ten Scientific and Technological Advances of 2020. This marked the first time that artificial intelligence had been used to rapidly and accurately simulate protein models, with results comparable to those obtained from expensive, complex, and time-consuming laboratory experiments.
In a previous interview, Shi Yigong stated, “The spatial three-dimensional structures of individual proteins within the human proteome that could be predicted have largely been predicted by AlphaFold. Overall, the predictions are reliable and fairly accurate. This represents a remarkable historic achievement in humanity’s scientific journey to understand the natural world.”
China is also home to no shortage of AI companies conducting in-depth research into biotechnology. Artificial intelligence companiesTianrang XLab recently announced that its self-developed deep learning protein folding prediction platform, TRFold, achieved a score of 82.7/100 in internal testing based on the CASP14 protein test set, ranking second globally and trailing only AlphaFold2, which scored 91.1/100.
It is reported that when predicting protein chains based on 400 amino acids, TRFold takes a maximum of only 16 seconds. By adopting weight sharing to conserve computational resources, TRFold uses only 8 GPUs, compared to the 128 TPUs (approximately equivalent to 256 GPUs) required by AlphaFold2, achieving performance comparable to AlphaFold2 with lower computational power and higher efficiency.
This represents the best performance achieved to date among all publicly available protein structure prediction models in China, marking that the country’s capabilities in computational biology have reached the world’s top tier.
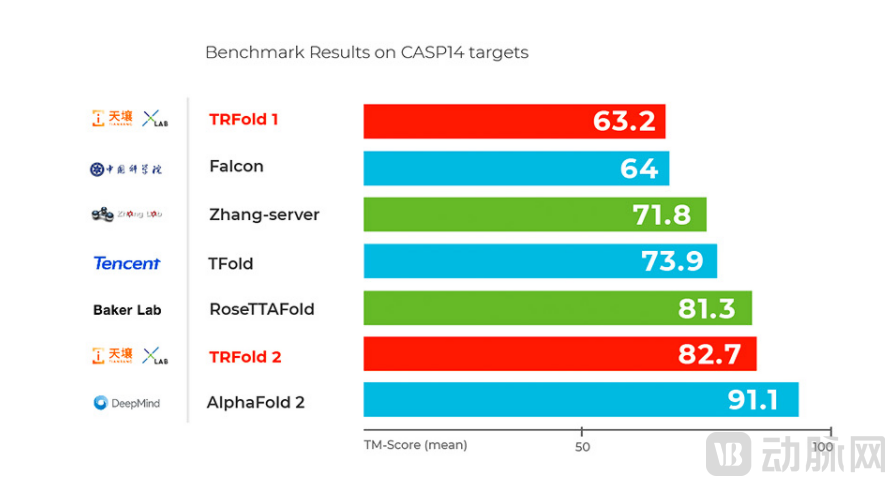
*RoseTTAFold results are from open-source predictions on GitHub; other data are from the official CASP website.
Held biennially, the CASP competition has become the most authoritative and prestigious event in the field of computational biology. Each iteration attracts numerous experts from diverse disciplines, including biophysics, computer science, high-energy physics, computational chemistry, and computational mathematics, earning it the reputation as the “Olympics of protein structure prediction.”
Traditional methods for observing protein structures mainly include three approaches: nuclear magnetic resonance (NMR), X-ray crystallography, and cryo-electron microscopy (cryo-EM). However, these methods often rely on extensive trial-and-error processes and expensive equipment, with each structure requiring years of research to determine. In contrast, the latest advancement in applying AI to protein structure prediction, AlphaFold2, can predict high-confidence protein structures within days or even minutes—a task that previously could take decades.
Four months after the open-source release of AlphaFold2, the iterative version of the Tianrang protein prediction model achieved the best performance in China during internal testing based on the CASP14 benchmark dataset, ranking second only to AlphaFold2.
According to Tianrang Information, AlphaFold2 represents a major breakthrough in protein structure prediction; however, the development of AI algorithms capable of addressing protein structure-function relationships and meeting the accuracy requirements for practical implementation has only just begun.TRFold possesses distinct advantages in model representation and training expertise, enabling it to address more complex, underlying challenges., such as protein-protein interactions. Compared with AlphaFold2, Tianrang has implemented numerous innovations and optimizations, offering significant advantages in model representation and computational efficiency, thereby demonstrating a pronounced late-mover advantage.
Tianrang is an innovative enterprise dedicated to research in general intelligence, committed to building a general artificial intelligence platform for complex systems. It aims to empower business scenarios with minimal cost and maximum speed, making intelligence as accessible as water, electricity, and gas. Currently, it is widely applied in scenarios such as urban operations, traffic management, financial insurance, and commercial retail.
Dr. Xue Guirong, Founder and CEOA leading scientist in the fields of artificial intelligence and big data, and a member of the Cloud Computing Expert Group under the Ministry of Science and Technology of China. Dr. Xue Guirong received his Ph.D. in Computer Science from Shanghai Jiao Tong University in 2006. From 2006 to 2009, he served as an Associate Professor and Distinguished Researcher in the Department of Computer Science at Shanghai Jiao Tong University. He is the first scientist in China to publish a paper at ACM SIGIR, the premier international conference in the field of search.
In 2009, Dr. Xue Guirong joined Alibaba Cloud, where he was responsible for developing the cloud-based Alibaba search engine (Shenma Search), supporting search and recommendation services for hundreds of millions of websites within Alibaba’s search framework. From 2013 to 2016, he served as Head of the Alimama Big Data Center and Chief Data Scientist at Alimama. He led the team in building the Damopan data management platform (DMP). Over this three-year period, daily advertising revenue grew from over RMB 10 million to more than RMB 80 million.
Dr. Xue Guirong has published over 70 papers at top-tier international conferences and in leading journals, holds more than ten patents, and has accumulated over 9,000 citations.
The team responsible for the TRFold project is called Tianrang XLab. Established in 2019, it primarily focuses on innovative fields. Its core members include doctoral candidates in biological computing, physics, mathematics, and related disciplines from top universities worldwide, fostering a strong culture of innovation.Over the past two and a half years, the XLab team has mastered core technologies in protein folding, enabling its participation in the most cutting-edge areas of international biomedicine., Tianrang has already crossed the technical threshold, enabling targeted R&D and application tailored to different scenarios in the future.
According to Dr. Xue Guirong, “Traditionally, a score above 90 indicates minimal deviation from laboratory-predicted results. Currently, TRFold has achieved relatively strong performance based on a smaller dataset, and further iterations are planned to push the score above 90. With technological breakthroughs, more application scenarios will emerge.”
With AlphaFold having already achieved tremendous success and been open-sourced, why still enter the field of protein structure prediction to develop a proprietary algorithm? Tianrang has its own perspective on this matter. Dr. Xue Guirong stated, “The success of AlphaFold2 represents a major breakthrough in protein structure prediction; however, the development of AI algorithms that address protein structure–function relationships and meet the accuracy requirements for practical real-world applications has only just begun. Without experience in model training, or without the capability to reproduce results comparable to those of AlphaFold2, it is impossible to leverage this technology to tackle more profound scientific challenges.”
For instance, AlphaFold-Multimer, released by the DeepMind team in October to predict proteins and protein–protein interactions, was developed by making minor adjustments to AlphaFold 2 and performing de novo training on protein complex structures to predict inter-protein relationships. Such in-depth research necessitates the capability to develop underlying algorithms independently for genuine application in the field of biology.
“Tianrang’s TRFold was independently developed with full consideration for downstream applications. For instance, our model platform offers different versions tailored to specific scenarios: the end-to-end version is designed for rapid structure generation, while the segmented version is used for large-scale calculation of distances between amino acids in proteins. Furthermore, extensibility and future research needs were thoroughly taken into account during development,” said Xue Guirong.
Dr. Miao Hongjiang, Project Leader of the Tianrang Protein Folding Initiative“It actually raised the barrier to entry by open-sourcing AlphaFold 2. Without the prior exploratory work, it would be impossible to quantify the advantages of its methodology or learn from its most valuable innovations in thinking. Furthermore, AlphaFold 2 did not release its training code, meaning that even if you download its source code, you can only predict single-protein structures. Rather than focusing solely on prediction, Tianrang places greater emphasis on the practical implementation of this technology. Therefore, we must build a proprietary algorithm from scratch to proceed with our subsequent work.”
Xue Guirong stated,Structural simulation of single proteins is just the beginning; based on the current TRFold, there are many directions for further exploration., such as simulating the interactions between proteins and their complexes (including small molecules, peptides, other proteins, etc.). Currently, a well-defined research direction is to further deepen the simulation of protein-protein interactions. Based on these interactions, several promising avenues include constructing large-scale interaction network maps, target discovery, simulating mutant protein structures, modeling post-translationally modified protein structures, GPCR simulations, and antibody simulations.
The company also revealed that its upcoming focus will be to leverage current whole-proteome co-evolutionary analysis to establish precise maps of protein–protein interactions, and to identify novel, precise therapeutic approaches for diseases by studying these interactions. Meanwhile, it aims to enhance the accuracy and success rate of protein design and explore new methods for the research, development, and design of proteins.
With the self-developed TRFold, Tianrang aims to help humanity build its own network of protein interactions, thereby making genuine contributions to disease treatment, drug development, and related fields.