Legal Considerations in Clinical Trials, Regulatory Approval, and Commercialization of Surgical Robots
Editor’s Note: This article is from AnJie Law Firm, authored by Cai Hang, Zhang Yin, and Wu Rongrong. Republished with permission by VCBeat.
"The razor's edge is sharp and difficult to cross." -- William Somerset Maugham
Surgical Robots: Once Hailed as Technological Innovation on the Tip of a ScalpelSurgical robots are typically defined as large-scale medical devices that integrate cutting-edge multidisciplinary technologies, including medicine, biomechanics, mechanics, computer science, and microelectronics. By leveraging a console, imaging systems, and robotic arms, they assist surgeons in performing precise control and completing complex surgical procedures through minimally invasive techniques. Compared with open surgery and traditional minimally invasive surgery, surgical robots offer numerous advantages. First, they effectively reduce the surface area of surgical incisions, enabling faster postoperative recovery for patients and reducing the incidence of postoperative complications. Second, the flexible robotic arms enhance surgical precision, ensuring the stability of surgical outcomes. Finally, surgical robots reduce the physical exertion required of surgeons, shorten the learning curve for physicians, and minimize patients’ exposure to radiation.
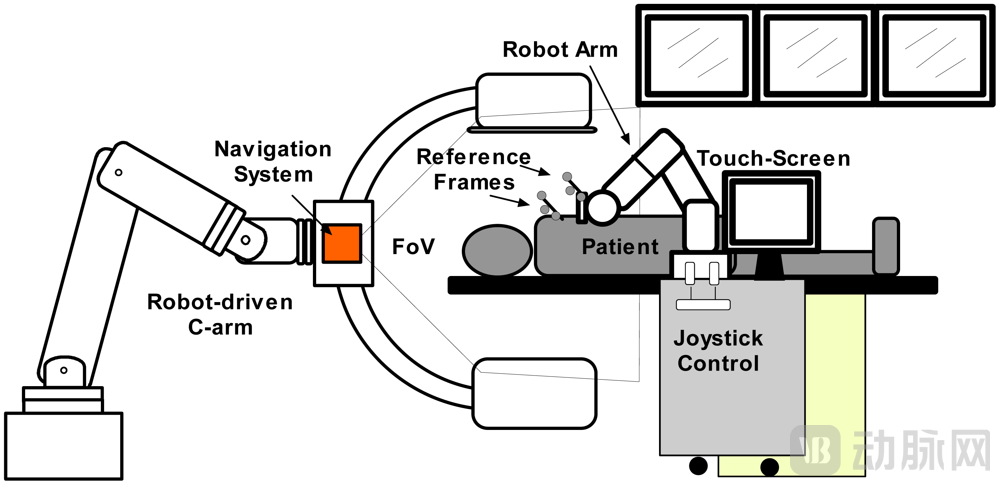
Surgical Robot System Diagram[1]
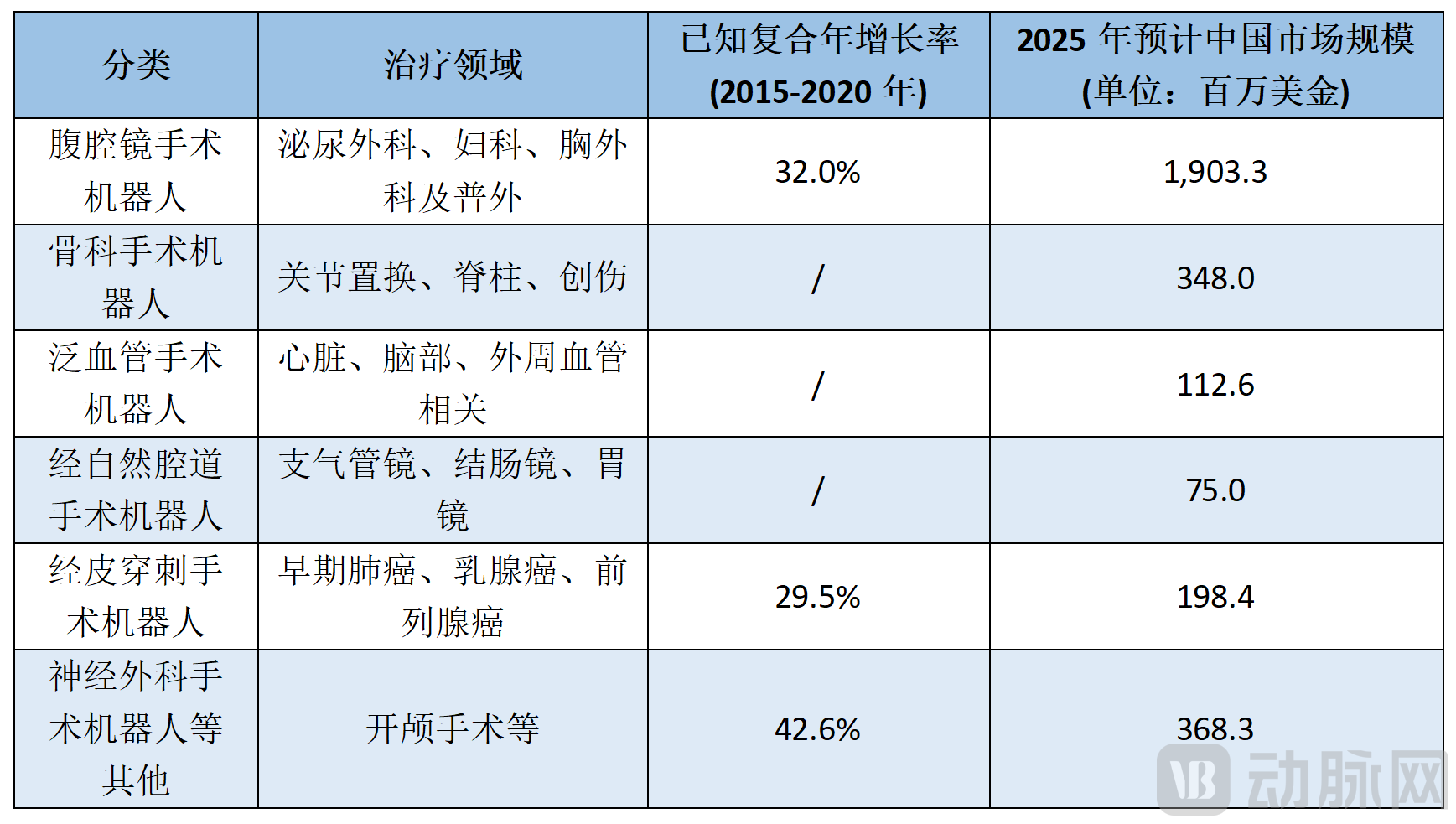
The global surgical robotics industry is experiencing rapid growth. By the end of 2021, more than ten surgical robotic products from foreign companies such as Intuitive Surgical, Stryker, and Medtronic had received market approval from the National Medical Products Administration (“NMPA”). Meanwhile, domestically produced surgical robots have also entered the market, with representative products including: (1) a product approved by the China Food and Drug Administration in 2016[2]Approved for market launch, developed by TinaviOrthopedic Surgical Navigation and Positioning Robot (TiJi), (2) approved for market launch by the NMPA in October 2021 and developed by WeigaoLaparoscopic Surgical Equipment (Miaoshou-S), and (3) approved for market launch by the NMPA in January 2022, developed by MicroPort MedBotEndoscopic Surgical Robot (Toumai). Compared with the 13.3% market penetration rate of laparoscopic robots in the United States in 2020, the penetration rate in China was only 0.51%.[3], therefore, the Chinese surgical robot market has just entered the development and introduction phase, with broad market space. According to Frost & Sullivan statistics[4], the following are the sales forecasts for surgical robots in China's market across various sub-sectors:
According to the research report published by Fosun Hengli[5], taking laparoscopic surgical robots as an example, the core technologies involved in surgical robots are shown in the figure below:
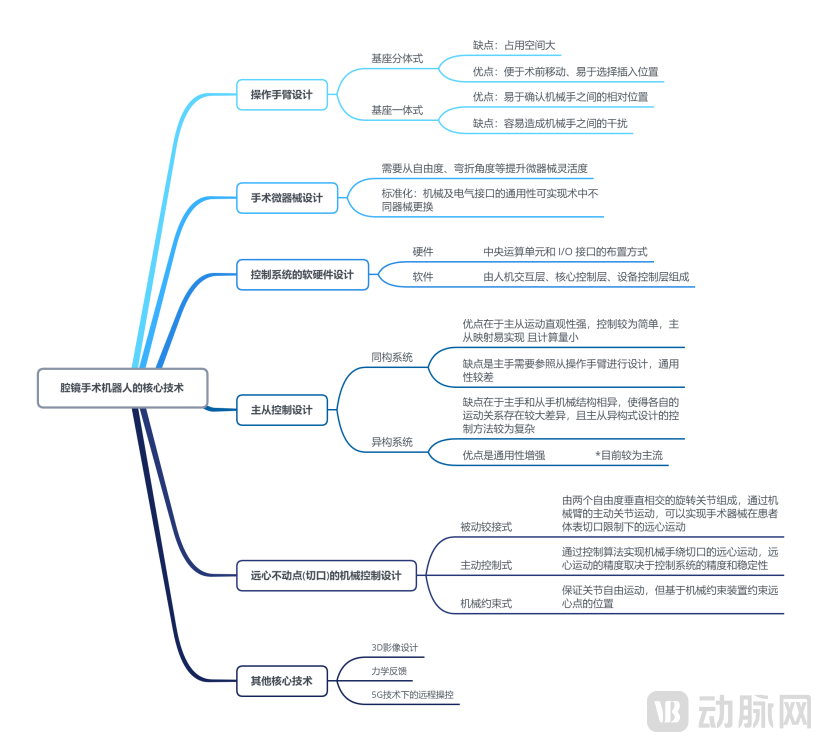
Diagram of Core Technologies for Laparoscopic Surgical Robots
As shown in the figure above, surgical robots involve a variety of technologies and present high technical barriers. Although domestically developed surgical robot products in China have not yet fully reached the same level as mature foreign counterparts such as the da Vinci system in terms of patent accumulation and clinical surgical outcomes, Chinese-made laparoscopic and orthopedic surgical robots have initially mastered key underlying core technologies and possess certain competitive advantages in novel functionalities such as haptic feedback and remote operation.
On the other hand, looking ahead, surgical robots can integrate artificial intelligence (AI) technologies. In addition to making surgeries more precise and efficient, this integration will transform surgical actions from qualitative maneuvers into quantifiable, standardized operational procedures and data information, thereby enabling the digitalization and intelligentization of surgical procedures. Specifically, various electrical signals collected by robotic arms and micro-instruments can be analyzed and optimized using large-scale datasets. This process, in turn, can further refine surgical workflows. Through iterative learning powered by AI, AI-enabled surgical robots are expected to achieve capabilities such as AI-assisted intraoperative guidance and even fully autonomous AI-driven operations. In the future, AI surgical robots may evolve into an integrated interactive carrier and output platform that connects intelligent diagnostics, preoperative planning, intraoperative guidance, and postoperative analysis.
1. Regulatory Submission and Registration Process for the Product as a Medical Device
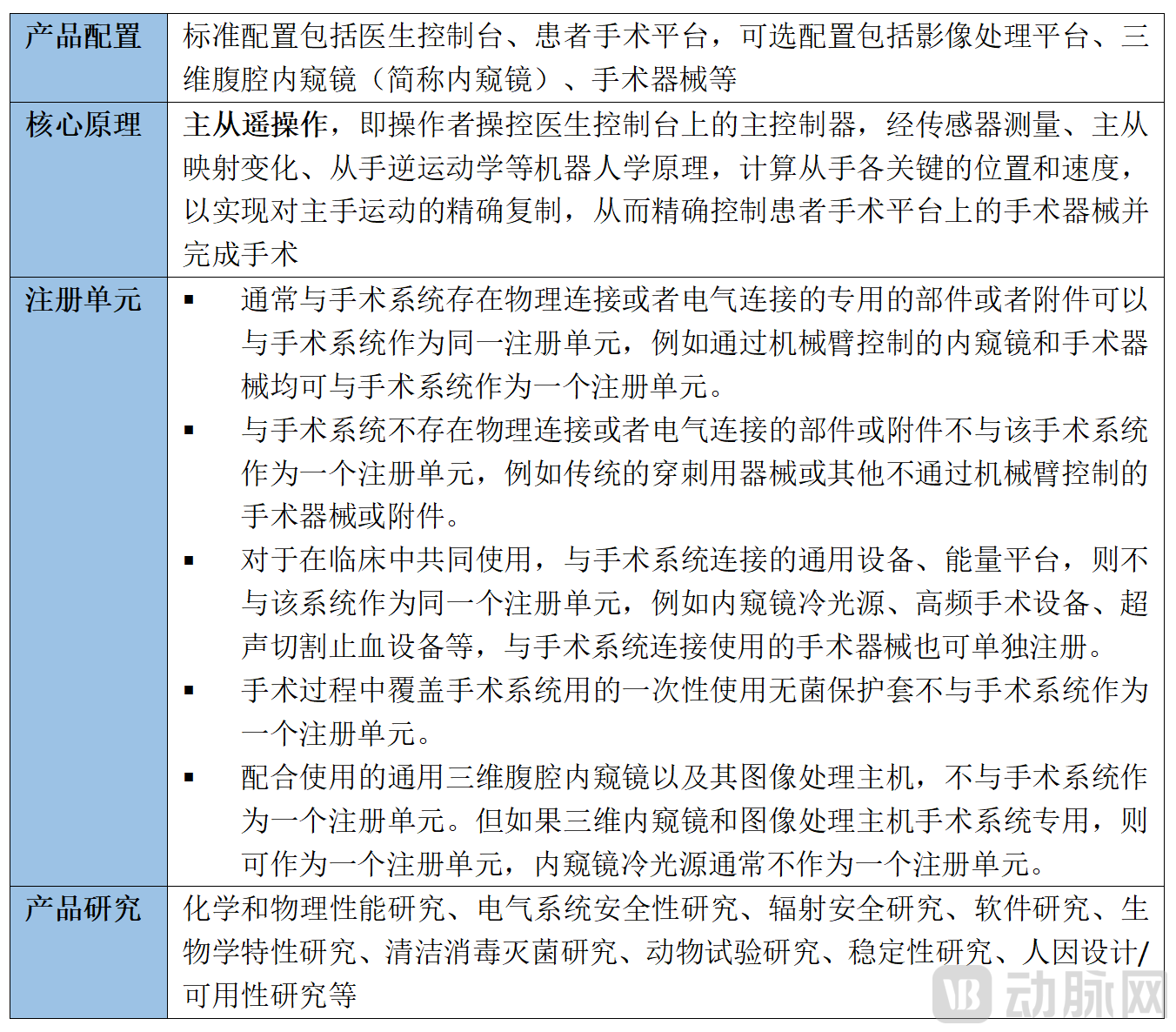
Surgical robots differ from traditional medical devices in that their complex structures and modular systems constitute an aggregation of multiple traditional medical devices, making it difficult to directly determine their classification based on the existing Medical Device Classification Catalog. According to the Regulations on the Supervision and Administration of Medical Devices, for newly developed medical devices not yet included in the classification catalog, applicants may either directly apply for product registration in accordance with the provisions for Class III medical device product registration, or determine the product category based on classification rules and apply to the National Medical Products Administration (NMPA) for category confirmation, followed by applying for product registration or filing as required. Surgical robots themselves are classified as active surgical instruments and implantable medical devices, posing a high risk to the human body. Under the currently effective Medical Device Classification Catalog, most items in the catalog of active surgical instruments, with few exceptions[6], are classified as Class III medical devices. Currently, all surgical robots that have been launched and put into use within China are regulated as Class III medical devices. In addition, according to the Technical Review Points for Laparoscopic Endoscopic Surgical Systems issued by the Center for Medical Device Evaluation (CMDE) of the National Medical Products Administration,“Key Points for Review”), laparoscopic surgical systems are explicitly regulated as Class III medical devices. Other important matters specified in the review guidelines are as follows:
As surgical robots are large-scale medical devices that integrate multiple cutting-edge technologies, a single product may incorporate and utilize numerous patented technologies. These advanced and innovative characteristics align perfectly with the national objective of encouraging research and innovation in medical devices and promoting high-quality development of the medical device industry. Therefore, such products can be registered and declared as innovative medical devices. Provided that the eligibility criteria for innovative medical devices are met, applicants may utilize the special review channel for innovative medical devices to enjoy priority processing, without lowering standards or reducing procedural requirements. For the applicable conditions of the special review procedure for innovative medical devices, please refer to Part (II) of this series, “Legal Issues Concerning the Application of Brain-Computer Interface Technology in Medical Scenarios.” The application process for the special review procedure for innovative medical devices is illustrated in the figure below:
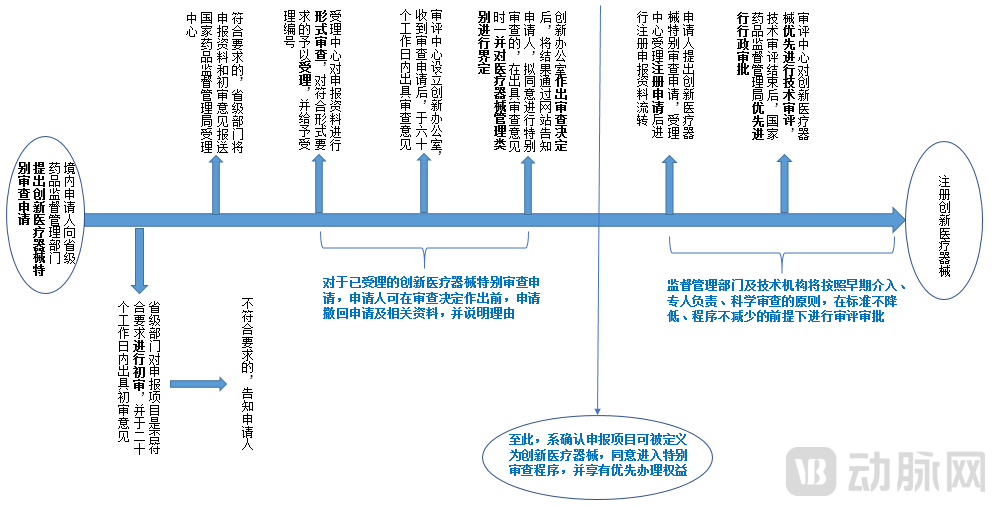
Flowchart of the Application Process for Innovative Medical Devices[7]
For imported surgical robot products intended for sale in the Chinese market, overseas enterprises must not only fulfill their obligations as registration applicants by submitting the requisite registration materials and supporting documentation to the National Medical Products Administration (NMPA), but also designate a domestic corporate entity as their local agent to submit these materials to the NMPA. The domestic agent shall further assist the overseas registrant in fulfilling the following obligations: establishing a quality management system appropriate for the product and ensuring its effective operation; formulating post-market research and risk control plans and ensuring their effective implementation; conducting adverse event monitoring and re-evaluation in accordance with the law; establishing and implementing product traceability and recall systems; and complying with other obligations stipulated by the drug regulatory department under the State Council. Notably, on June 29, 2022, the NMPA issued the Implementation Plan for Supporting Medical Device Registrants from Hong Kong and Macao to Produce Medical Devices in Nine Mainland Cities of the Greater Bay Area, marking the pilot launch and gradual expansion of the cross-border contract manufacturing system for imported medical device registrants in China. These favorable regulatory policies related to the market approval and production of imported medical devices are conducive to promoting cross-border cooperation in the field of surgical robots.
2. Protection of Participants' Personal Information During Clinical Trials
From a normative perspective, the Good Clinical Practice for Medical Device Trials, the Declaration of Helsinki of the World Medical Association, and the Personal Information Protection Law ("Personal Information Protection Law") all explicitly require sponsors to assume relevant obligations regarding the processing of subjects’ personal information and the protection of their right to know and right to privacy. R&D enterprises should pay particular attention to the following legal issues related to personal information processing:
(1) Separate consent must be obtained from individuals before processing sensitive personal information
The Personal Information Protection Law explicitly stipulates the processing of sensitive personal information.[8]Separate consent must be obtained from the individual. Prior to processing such sensitive personal information, the individual shall be accurately informed, in clear and understandable language, of elements including the name and contact details of the personal information processor, the methods and purposes of processing, the retention period for the information, the necessity of processing sensitive personal information, and the impact on the individual’s rights and interests. In cases involving multi-regional clinical trials or international collaborative clinical trials, consideration must be given to whether the personal information of domestic participants may be shared with overseas entities. If cross-border transfer is required, the participant shall also be informed of matters such as the name or title and contact details of the overseas recipient, the purposes and methods of processing, the categories of personal information involved, and the means and procedures by which the individual may exercise their rights under the Personal Information Protection Law vis-à-vis the overseas recipient, and separate consent must be obtained from the individual.
Generally, the informed consent form signed by subjects prior to their participation in clinical trials contains specific clauses on personal information protection. These clauses specify the types of personal information that may be collected from subjects, as well as the storage locations, access rights, and scope of sharing for such collected personal information. Furthermore, the clinical trial institution or the investigator team provides detailed explanations before the informed consent form is signed. We understand that the aforementioned practices (including the subject’s signing of the informed consent form after being fully informed) may, to some extent, be regarded as an effective means of obtaining the subject’s separate consent for the processing of their personal information. However, whether these practices necessarily meet the compliance requirements for obtaining the subject’s separate consent for the processing of their sensitive personal information requires further determination based on the wording and scope of the specific personal information protection clauses within the informed consent form.
To effectively mitigate the risk of violating laws and regulations governing the processing of sensitive personal information, we hereby provide the following recommendations regarding the procedures for obtaining informed consent and the inclusion of special clauses on personal information protection for your reference:
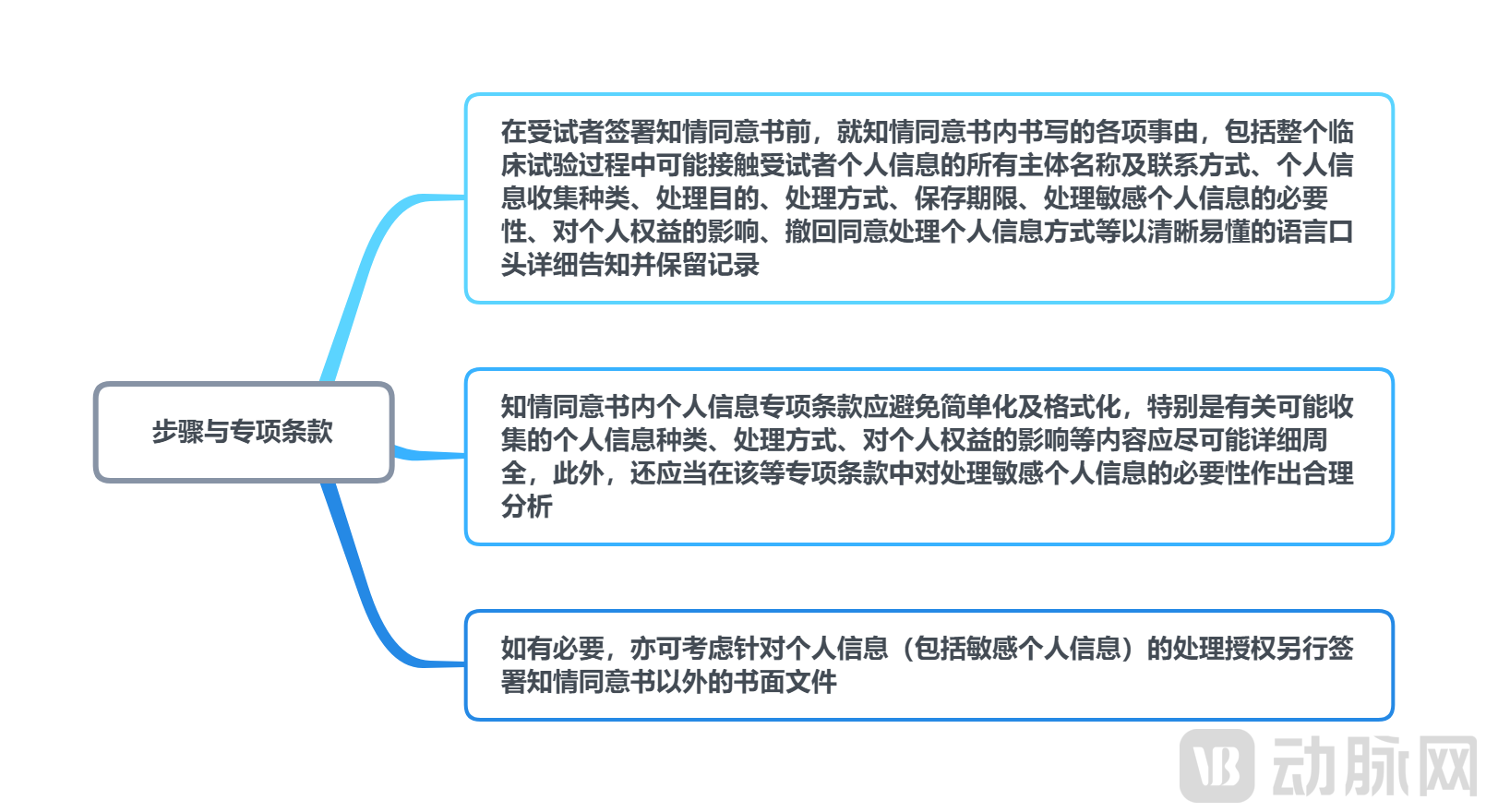
Illustrative Guide to Personal Information Protection Recommendations
(2) Assessment Obligations of Clinical Trial Sponsors as Personal Information Processors
Under the Personal Information Protection Law, clinical trial sponsors, as processors of sensitive personal information, may be involved in the following assessment activities:
● Security Assessment for the Cross-Border Transfer of a Specific Volume of Personal Information: In the Case of Multi-Regional Clinical Trials[9]The primary data analysis center is located overseas, which will involve the cross-border transfer of sensitive personal information of domestic trial subjects. Conducting international collaborative clinical trials may also entail cross-border data transfers. The volume of personal information transferred reaches the threshold stipulated by the national cyberspace administration authorities.[10]Personal information processors shall store personal information collected and generated within the territory of the People's Republic of China domestically. Where it is truly necessary to provide such information overseas, a security assessment organized by the national cyberspace administration authority must be passed. In other circumstances, compliance may be achieved either through personal information protection certification conducted by professional institutions or by concluding a contract with the overseas recipient that stipulates respective rights and obligations. It should be noted that prior to applying for the data cross-border transfer security assessment, clinical trial sponsors must conduct a self-assessment of data cross-border transfer risks and submit the self-assessment report to the cyberspace administration authority when filing for the security assessment. Furthermore, attention should be paid to whether human genetic resources may be involved.[11]The export of materials, if applicable, shall be subject to a joint application by both cooperating parties and approval by the State Council’s department responsible for science and technology administration. If the activity does not involve export but only collection, the type, quantity, and intended use of the human genetic resources to be utilized shall be filed with the State Council’s administrative department for science and technology prior to the initiation of clinical trials; failure to comply will result in substantial fines.[12]。
● Personal Information Protection Impact Assessment: In any of the following circumstances, personal information processors shall conduct a personal information protection impact assessment in advance and document the processing activities: a) processing sensitive personal information; b) using personal information for automated decision-making; c) entrusting the processing of personal information to third parties, providing personal information to other personal information processors, or disclosing personal information to the public; d) transferring personal information overseas; e) other personal information processing activities that have a significant impact on individuals’ rights and interests. We understand that, as sponsors of clinical trials, R&D enterprises, once they become entities processing sensitive personal information, are inherently obligated to carry out personal information protection impact assessments.[13], such reports and records of handling shall be retained for at least three years.
3. Key Considerations for Entrusting CROs and SMOs with Clinical Trials
As innovative medical devices, surgical robots entail a high-investment, long-cycle systematic race in their research and development (R&D) and regulatory approval for market launch. To ensure the smooth implementation of clinical studies and clinical trials, R&D enterprises often engage Contract Research Organizations (CROs) and Site Management Organizations (SMOs).[14]Third-party R&D assistance services delegate clinical trial operations management, clinical trial site management, and other clinical non-core R&D activities to CROs and/or SMOs for final implementation.
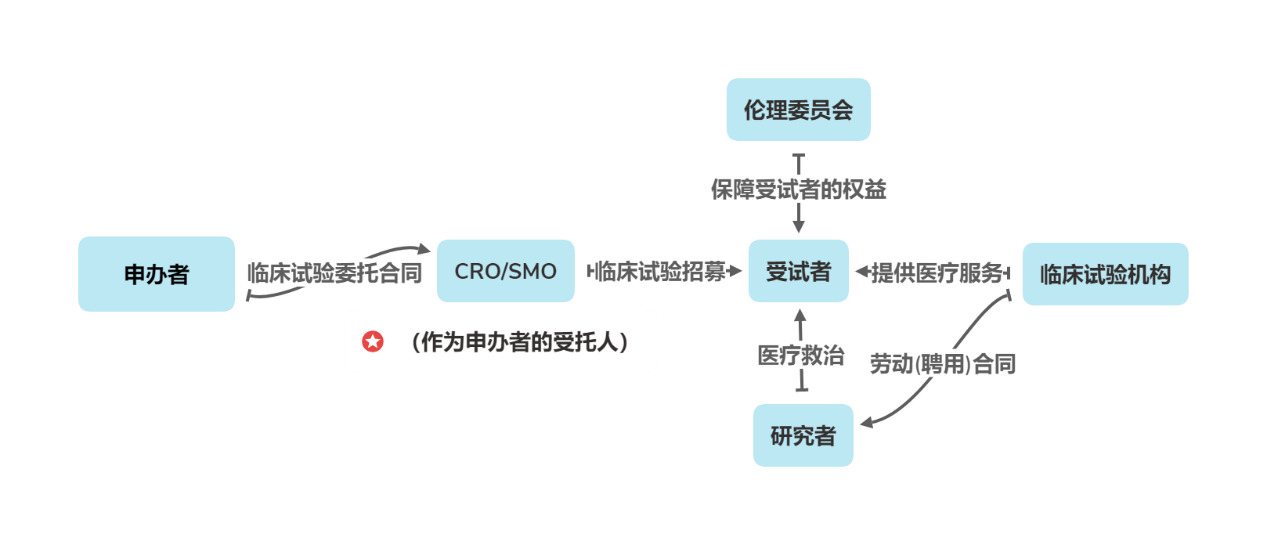 Diagram of Relationships Among Stakeholders in Medical Device Clinical Trials
Diagram of Relationships Among Stakeholders in Medical Device Clinical Trials
According to the Measures for the Registration and Filing of Drug Research Institutions (Trial) (“Filing Measures”), any institution engaged in research in China for the purpose of applying for clinical trials and marketing approval of drugs (explicitly including contract research organizations, or CROs) shall apply for registration and filing with the provincial-level drug regulatory authorities in accordance with such measures. However, in practice, certain regions have ceased to enforce the Filing Measures, and some have abolished such filing requirements altogether. Consequently, CROs conducting clinical trial-related service activities are unable to complete the registration and filing as drug research institutions with local drug regulatory authorities pursuant to the provisions of the Filing Measures. This is further supported by publicly available disclosure documents from listed companies in the CRO and site management organization (SMO) industries.[15], the issuer’s sponsor institution and legal counsel generally hold the view that the NMPA has not established market entry policies for the CRO and SMO industries, and that CRO and SMO enterprises are not currently required to obtain specific qualifications or licenses for the operation and site management services of clinical trials.
As highly specialized contract outsourcing services, CROs and SMOs command relatively high project service fees and involve complex scopes of work. Therefore, surgical robot R&D enterprises should pay attention to the following key points when signing clinical trial delegation contracts with CROs and SMOs:
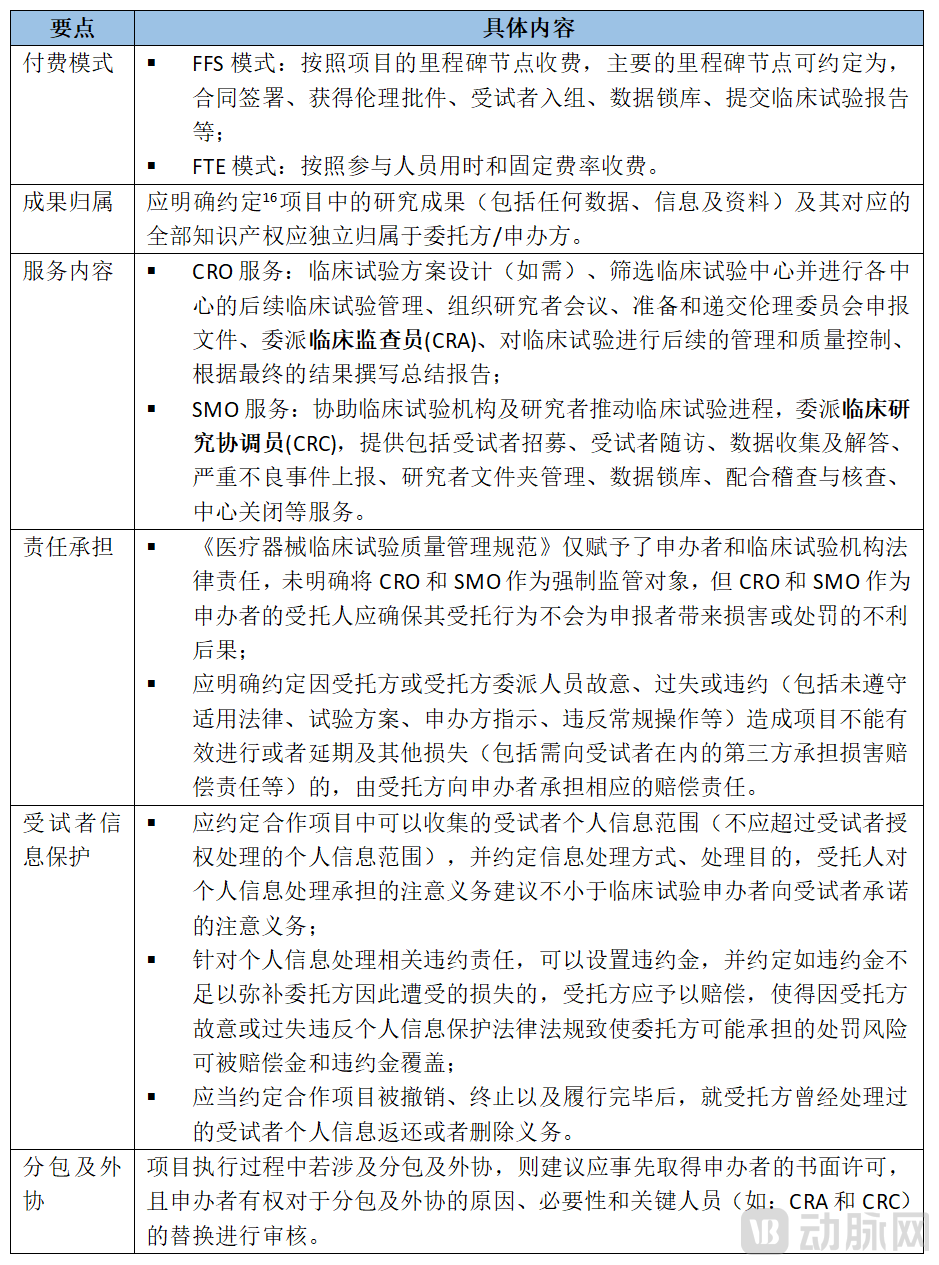
4. When selling surgical robots, which are classified as large-scale medical equipment, it is essential to verify whether the healthcare institution has obtained a configuration permit.
China implements a system of tiered and classified allocation planning and licensing management for the installation and use of large-scale medical equipment. Medical institutions intending to allocate products listed in the Catalogue for Licensing Management of Large-Scale Medical Equipment Configurations must apply to the health administrative authorities and obtain an Allocation License. According to the latest 2018 catalogue[17]“The Endoscopic Surgical Instrument Control System” surgical robot is classified as a Class B medical device, subject to allocation management by provincial health commissions. Specifically, medical institutions must obtain approval from the provincial health administrative department and secure an allocation permit prior to procurement. The general workflow for medical institutions when procuring large-scale medical equipment is illustrated in the figure below:
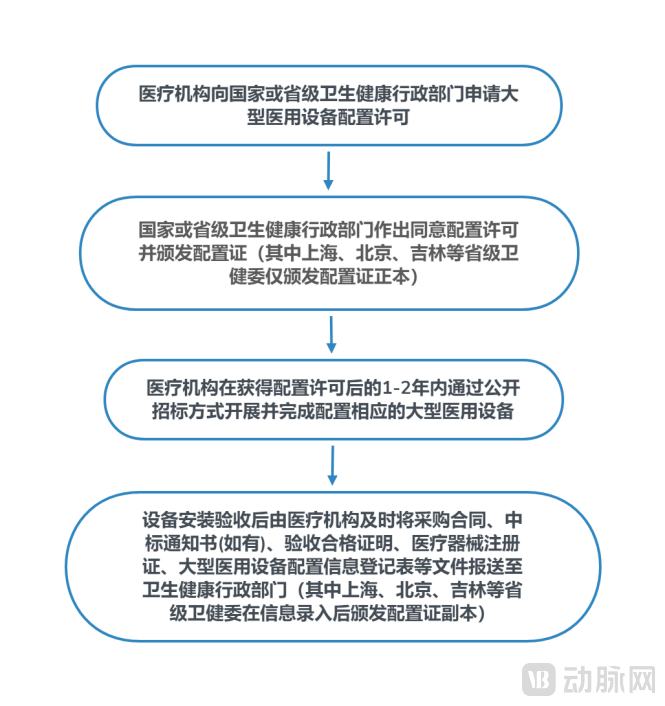
General Flowchart for the Procurement of Large-Scale Medical Equipment by Healthcare Institutions
Regulatory oversight of hospital procurement of large-scale medical equipment is being gradually relaxed. According to the "Implementation Plan for the 'Separation of Permits and Licenses' Reform in the Allocation of Large-Scale Medical Equipment in Socially Run Medical Institutions," issued by the National Health Commission on June 30, 2021: (1) The allocation permit for Class B large-scale medical equipment in private medical institutions shall be subject to a notification and commitment system, whereby applicants are informed at once of the conditions and required materials for obtaining the permit; if an applicant voluntarily commits to meeting the licensing conditions and submits the required materials accordingly, the permit decision shall be made on the spot; (2) For private medical institutions within pilot free trade zones, the management of Class B large-scale medical equipment allocation shall shift from approval-based to record-filing-based, exempt from restrictions imposed by large-scale medical equipment allocation plans.
Furthermore, in combination with the A-share IPO application materials of large-scale medical equipment enterprises obtained through public inquiries[18]Whether end customers hold configuration permits for the purchased equipment has become one of the legal issues focused on by stock exchanges and regulatory authorities during the review of initial public offerings (IPOs). Fortunately, in previous IPO cases, where the number of medical institutions among end customers that failed to provide configuration permits and the corresponding revenue accounted for a relatively low proportion, such issues did not constitute substantial obstacles to the issuer’s listing. Furthermore, the issuer’s counsel opined that the end customers’ failure to obtain configuration permits constitutes a violation of administrative mandatory provisions rather than validity-related mandatory provisions. In light of judicial precedents, the lack of configuration permits does not affect the valid formation of sales contracts entered into between the issuer and end customers or distributors.
In subsequent market-based sales, surgical robot manufacturers should carefully review and retain records of end customers’ configuration permits, while actively expanding their diversified client base to include public hospitals and private hospitals.
We have published a series of articles focusing on AI-powered rehabilitation robots, brain-computer interface (BCI) devices, and surgical robots. Artificial intelligence is driving the continuous launch of innovative medical devices. On one hand, with the support of various policies from the National Medical Products Administration (NMPA), the approval rules and review criteria for innovative devices have become increasingly clear. On the other hand, while R&D enterprises and industry investors focus on technological innovation, they should also pay attention to compliance requirements across various aspects, including upstream and downstream supply chain relationships, core corporate technologies, clinical trials, import and export controls, and data protection. We hope this series of articles will serve as a valuable reference and benefit professionals in China’s intelligent medical device industry.
Author:

AnJie Law Firm’s Healthcare Team
Contact QR Code

Mr. Cai Hang has specialized in investment and financing services in the healthcare, TMT, and artificial intelligence sectors for over a decade, wielding significant influence in China’s venture capital legal services landscape. *China Business Law Journal* named him one of the “100 Legal Elites in China,” recognizing him as one of the country’s most outstanding commercial lawyers. He has also been repeatedly recommended by leading legal ranking agencies such as The Legal 500 and Legalband in the fields of TMT and venture capital. In addition to venture capital work, he is highly proficient in mergers and acquisitions and capital markets practices. Mr. Cai serves as Managing Partner of AnJie Broad Law Firm’s Shanghai office. Email: caihang@anjielaw.com

Ms. Zhang Yin has represented renowned domestic and international funds in numerous investment and M&A transactions, while also providing legal services for corporate clients’ financing and investment activities. Her practice spans industries including healthcare, biopharmaceuticals, artificial intelligence, entertainment and media, and tourism. Legalband recognized her as one of the “Top 30 Rising Stars in China’s Legal Profession 2021.” In the field of big health compliance, Ms. Zhang has assisted healthcare companies in deeply engaging with internet hospital compliance matters and has represented clients in completing acquisitions of multiple medical institutions and healthcare internet enterprises. Email: zhangyin@anjielaw.com

Wu Rongrong, LawyerEmail: wurongrong@anjielaw.comMs. Wu primarily specializes in private equity investment and financing, as well as mergers, acquisitions, and restructurings. She has participated in numerous domestic and cross-border private equity transactions for renowned investment institutions and enterprises, covering industries such as culture and entertainment, food and beverage services, gaming, software, SaaS, and pharmaceuticals.
*References
[1]Tovar-Arriaga, Saúl, José Emilio Vargas, Juan M. Ramos, Marco A. Aceves, Efren Gorrostieta, and Willi A. Kalender. 2012. "A Fully Sensorized Cooperative Robotic System for Surgical Interventions" Sensors 12, no. 7: 9423-9447. https://doi.org/10.3390/s120709423.
[2] In March 2018, the Plan for Reforming State Council Institutions, reviewed and approved at the First Session of the 13th National People's Congress, decided to abolish the China Food and Drug Administration (CFDA) and establish the National Medical Products Administration (NMPA).
[3] HJ Circle: “Research Report on China’s Surgical Robotics Industry (Overview),” released in March 2022.
[4] For details, please refer to the prospectus of MicroPort MedBot (02252.HK).
[5] He Shuanglin (Fuxing Hengli Research Department): “In-Depth Report on China’s Laparoscopic Surgical Robot and Orthopedic Surgical Robot Industries,” published in December 2021.
[6] The following items in active surgical instruments are not classified as Class III medical devices: accessories for ultrasonic surgical equipment; medical laser optical fibers; high-frequency surgical equipment used for coagulation, denaturation, and/or necrosis of corresponding tissues during superficial surgeries in dermatology, otolaryngology, gynecology, and proctology; electric staplers used for dissection, resection, and/or anastomosis of internal organs and tissues, suitable for various open or minimally invasive surgeries, etc.
[7] In the figure, (1) “Acceptance Center” refers to the Administrative Matters Acceptance Service and Complaints & Reporting Center; (2) “Evaluation Center” refers to the Center for Medical Device Technical Evaluation; (3) “Innovation Office” refers to the Innovation Medical Device Review Office.
[8] The information of subjects collected and recorded in clinical trials covers names, ages, past medical records, various laboratory indicators, examination results, surgical plans, etc. Once such personal data related to medical health is leaked or illegally used, it is easy to cause infringement of the subject's human dignity or harm to personal and property safety, which belongs to sensitive personal information.
[9] When multicenter clinical trials are conducted in different countries or regions, they are referred to as multi-regional clinical trials.
[10] See Article 4 of the Measures for the Security Assessment of Outbound Data Transfers (effective September 1, 2022) for details.
[11] Including human genetic resource materials and human genetic resource information. Human genetic resource materials refer to genetic materials such as organs, tissues, and cells that contain human genetic material, including the human genome and genes. Human genetic resource information refers to data and other informational materials generated from the use of human genetic resource materials.
[12] The science and technology administrative department of the State Council shall order the cessation of illegal activities, confiscate the illegally collected or preserved human genetic resources and any illegal gains, and impose a fine of not less than RMB 500,000 but not more than RMB 5 million; where the illegal gains exceed RMB 1 million, a fine of not less than five times but not more than ten times the illegal gains shall be imposed.
[13] A personal information protection impact assessment shall include the following contents: (1) whether the purposes and methods of processing personal information are lawful, legitimate, and necessary; (2) the impact on individuals’ rights and interests and security risks; (3) whether the protective measures adopted are lawful, effective, and commensurate with the level of risk.
[14] SMOs are generally regarded as a specialized segment of CROs, with a greater focus on the aspect of clinical site management.
[15] According to the prospectuses and legal opinions submitted by Northgate (301333) and Puruisi (301257) during their IPO applications, both the sponsoring institutions and lawyers of the issuers hold the view that the NMPA has not yet implemented approval or industry access policies for CRO/SMO companies.
[16] According to Article 859 of the Civil Code, unless otherwise provided by law or agreed upon by the parties, the right to apply for a patent for an invention-creation resulting from commissioned development shall belong to the researcher-developer. Where the researcher-developer obtains the patent right, the commissioning party may exploit such patent in accordance with the law. In light of this, if the terms of the commissioned contract are unclear, it will create obstacles for the sponsor to claim ownership of the research results.
[17] According to the provisions of the "Catalogue for the Administration of Licensing for the Allocation of Large-Scale Medical Equipment (2018)," in addition to the 10 explicitly listed categories of equipment, there are catch-all provisions based on price ranges. Large-scale medical devices with a unit price of RMB 30 million or USD 4 million or more for initial allocation are classified as Class A devices, while those with an initial allocation price ranging from RMB 10 million to RMB 30 million are classified as Class B devices.
[18] According to the legal opinion submitted by United Imaging Healthcare (688271) during its IPO application, the issuer’s counsel analyzed and addressed the regulatory concerns raised by the China Securities Regulatory Commission (CSRC) regarding the legal risks associated with end customers failing to obtain configuration licenses.