Companion Diagnostics Industry Report 2022: From 'Shovel Buyers' to 'Shovel Makers', Ushering in a Billion-Dollar Market Competition
Preface
In recent years, with the rise of targeted therapy and immunotherapy, precision medicine has gradually come into the spotlight. Targeted drugs and immunotherapeutic agents have created an anti-cancer myth with unprecedented precision. Precise medications require precise diagnostics; only after determining the molecular subtype can targeted treatment be administered, giving rise to companion diagnostics.
Nowadays, companion diagnostics have become an indispensable testing step prior to medication administration, representing a core subsector of in vitro diagnostics that has entered the commercialization phase. With regulatory policies standardizing companion diagnostics, a proliferation of technological approaches, and differentiated business models, the market landscape for companion diagnostics has already taken shape.
New vitality stems from innovations in technology and business models. Technologically, mainstream techniques such as PCR, IHC, FISH, and NGS have undergone varying degrees of upgrading, while emerging technologies like single-cell analysis, mass spectrometry, and multi-omics are demonstrating new potential, with AI enabling a closed-loop ecosystem. What “gold standards” must be met for successful clinical application of these technologies?
From a business model perspective, LDTs are beginning to be implemented through hospital pilots, overseas expansion is showing significant growth momentum, there have been few attempts to extend services to primary care institutions, co-built laboratories remain highly controversial, and joint development with pharmaceutical companies is gradually gaining traction. What are the underlying development logics for each of these models? And what will be their future trajectories?
Guided by these questions, we engaged in discussions with more than 15 companies and nearly 30 experts. The “2022 Companion Diagnostics Industry Research Report” primarily explores the application of companion diagnostics in oncology, addresses the aforementioned questions, and draws the following conclusions regarding technology, business models, and reimbursement:
Technologically, mainstream and emerging forces advance in tandem.PCR and gene sequencing have advanced to third-generation technologies, mIHC enables multiplex labeling, and FISH hybridization has become faster. Single-cell technologies provide analytical insights at the cellular level, while mass spectrometry serves as a critical analytical technique in the field of biomacromolecule research. In the future, multi-technology and multi-omics platforms will jointly drive industry development, with AI enabling closed-loop systems. For successful clinical application, technologies must not only meet basic requirements such as sensitivity and stability but also be evaluated for their application potential across five dimensions: standardization, ease of use, throughput, technical costs, and personnel costs.
In terms of models, LDTs face varying thresholds and levels of acceptance for hospital entry.In China, barriers may be established by requiring Laboratory Developed Tests (LDTs) to gain hospital access rather than through an approved regulatory pathway. Regardless of whether the model involves co-built laboratories or collaborations with pilot hospitals, hospital access for LDTs remains a significant barrier. Under such barriers, product acceptance inevitably varies; LDT products piloted in large tertiary Grade A hospitals will undoubtedly face lower approval hurdles compared to those piloted in secondary hospitals or third-party testing institutions. Meanwhile,Where LDT products are implemented will become a new benchmark. In terms of regulatory complexity, the pilot hospitals for LDTs at this stage are more likely to be large public tertiary hospitals.
In terms of payment, medical insurance coverage is accelerating, and PCR tests may be the first to undergo centralized procurement.Based on the historical trends of oncology genetic testing projects and the response from the National Healthcare Security Administration (NHSA) on October 12, 2022, regarding their inclusion in medical insurance and volume-based procurement (VBP), we believe that, in the long term, including oncology genetic testing in medical insurance will facilitate the implementation of companion diagnostic projects in hospitals. In the short term, due to the high costs of next-generation sequencing (NGS) and the substantial investment required for large-panel development, it is unlikely to be covered by medical insurance. Meanwhile, polymerase chain reaction (PCR) technology, being widely applied, more mature, and lower in cost, may be the first to undergo VBP.
Policies Are Gradually Being Standardized, Unveiling a Blue-Ocean Market Worth Over RMB 10 Billion
Policy: China Enters Adjustment PhasePhase, will continue to standardize
Companion diagnostics originated from precision medicine and have been widely adopted globally in recent years. The United States has the earliest inception and the most mature development in the companion diagnostics industry. From the perspective of U.S. regulatory policies, the co-development of drugs and test kits serves as the foundational principle. Currently, the companion diagnostics industry in the United States has reached a mature stage.
Development Stages of U.S. Companion Diagnostic Policies
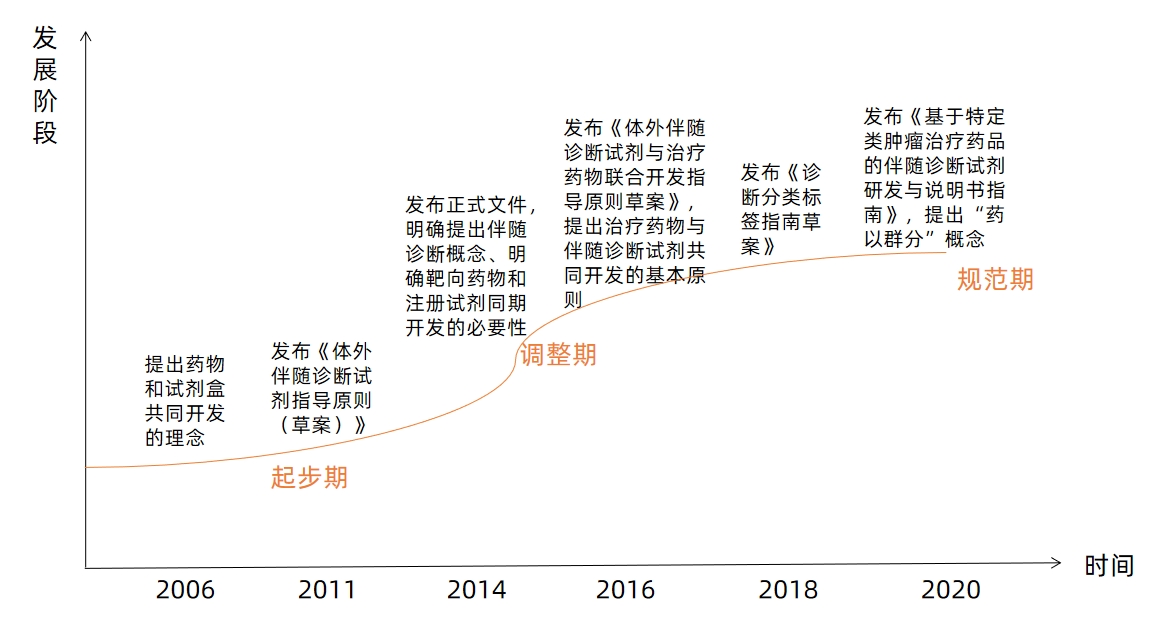
Image source: VCBeat
Based on the new regulations introduced in 2021 and 2022, it is evident that China has also begun to encourage the co-development of drugs and companion diagnostics. Overall, China’s companion diagnostics policy has entered an adjustment phase, while the market is experiencing rapid growth.
Development Stages of China's Companion Diagnostics Policies

Image source: VCBeat
Financing: 53 companies secured funding in the past three years, with Series B being the most common in the latest round
According to incomplete statistics, from 2020 to the present, a total of 53 companies involved in the field of companion diagnostics have secured financing. Among them, Series B is the most common latest funding round, accounting for 32.7%.
In addition, financing rounds over the past three years have shown significant differences in both technology and business models. Investments have primarily focused on emerging technologies such as single-cell analysis and multi-omics, as well as breakthroughs in mainstream technologies. In terms of business models, funded companies have largely adopted diversified operations rather than focusing solely on companion diagnostics. Currently, there are three large-scale companies in the companion diagnostics sector that have completed their initial public offerings.
Overall, the commercialization of companion diagnostics is relatively mature, with leading enterprises having seized the first-mover advantage. New technologies and business models are poised for emergence, creating a market landscape where large platforms coexist with specialized, niche players, and the competitive structure has already taken shape.
Market: Late Start, Rapid Growth, Huge Potential
According to Zhiyan Consulting, the global market size for companion diagnostics has been expanding year by year, reaching $3.76 billion in 2019, with a compound annual growth rate (CAGR) of 25.5% from 2016 to 2019. In terms of market growth rate, this sector remains in a phase of rapid development. Based on a projected growth rate of 25.5%, the global companion diagnostics market is estimated to reach $7.43 billion in 2022 and $14.69 billion in 2025.
In 2019, the market size of companion diagnostics in China reached RMB 2.72 billion, with the growth rate increasing year by year. The compound annual growth rate (CAGR) from 2016 to 2019 was 33.3%. Based on this 33.3% growth rate, the market size of companion diagnostics in China is estimated to reach RMB 6.44 billion in 2022 and RMB 15.26 billion in 2025.
Market Size of Global and China Companion Diagnostics
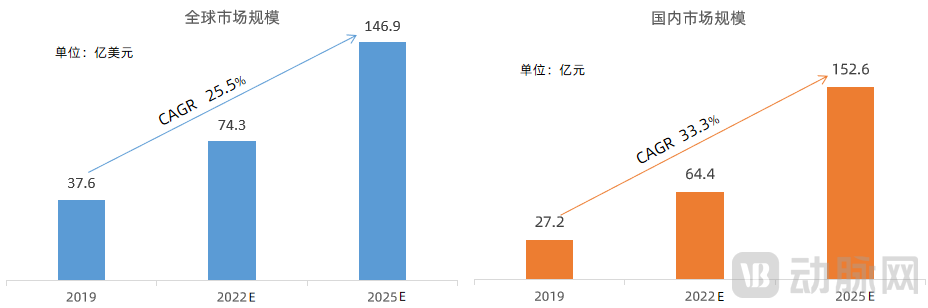
Data Source: Zhiyan Consulting, VCBeat
Four Major Technology Upgrades, Three Emerging Technologies Accelerating Adoption
Mainstream technologies have been upgraded, with PCR and NGS as the core modalities.
Currently, the most widely used technologies in the field of companion diagnostics are PCR andGene Sequencing,PCRand gene sequencing bothUnderwent two upgrades.
PCR advancements include second-generation quantitative real-time PCR (qPCR) and third-generation digital PCR (dPCR), although dPCRCompared with qPCR, it offers improvements in quantitative analysis, but the technology is not yet mature; currently, the most widely used method is qPCR。
Advancements in gene sequencing include next-generation sequencing (NGS) and third-generation single-molecule sequencing. Although third-generation single-molecule sequencing offers longer read lengths, it is still in the early stages of application. Currently, the most widely used technology isNGS。
IHC is limited in the number of biomarkers it can assess, whereas multiplex immunohistochemistry (mIHC) currently enables simultaneous staining for 7–9 markers. FISH assays are time-consuming; however, rapid FISH technologies have reduced hybridization time from a minimum of 8 hours to 2 hours. These techniques each have distinct characteristics and are complementary in clinical application.We have summarized the characteristics of these technologies as follows:
Comparison of Key Features Before and After the Upgrade of Mainstream Technologies

Image source: VCBeat
Based on the technologies used in currently approved companion diagnostic products, PCR-based reagents have received the most approvals. As of October 14, 2022, there were 55 approved PCR reagents, 16 NGS assays, 6 IHC assays, and 27 FISH assays for companion diagnostics; we have compiled statistics on these approval figures separately.
PAmong the reagents approved via the Clinical Review (CR) pathway, in addition to those intended for companion diagnostics, others are used for pharmacogenomics, auxiliary tumor diagnosis, and pathogenic microorganism testing. We have compiled statistics on products designated for companion diagnostics, as follows:
PCRStatistics on Approved Reagents for Companion Diagnostics by Technology
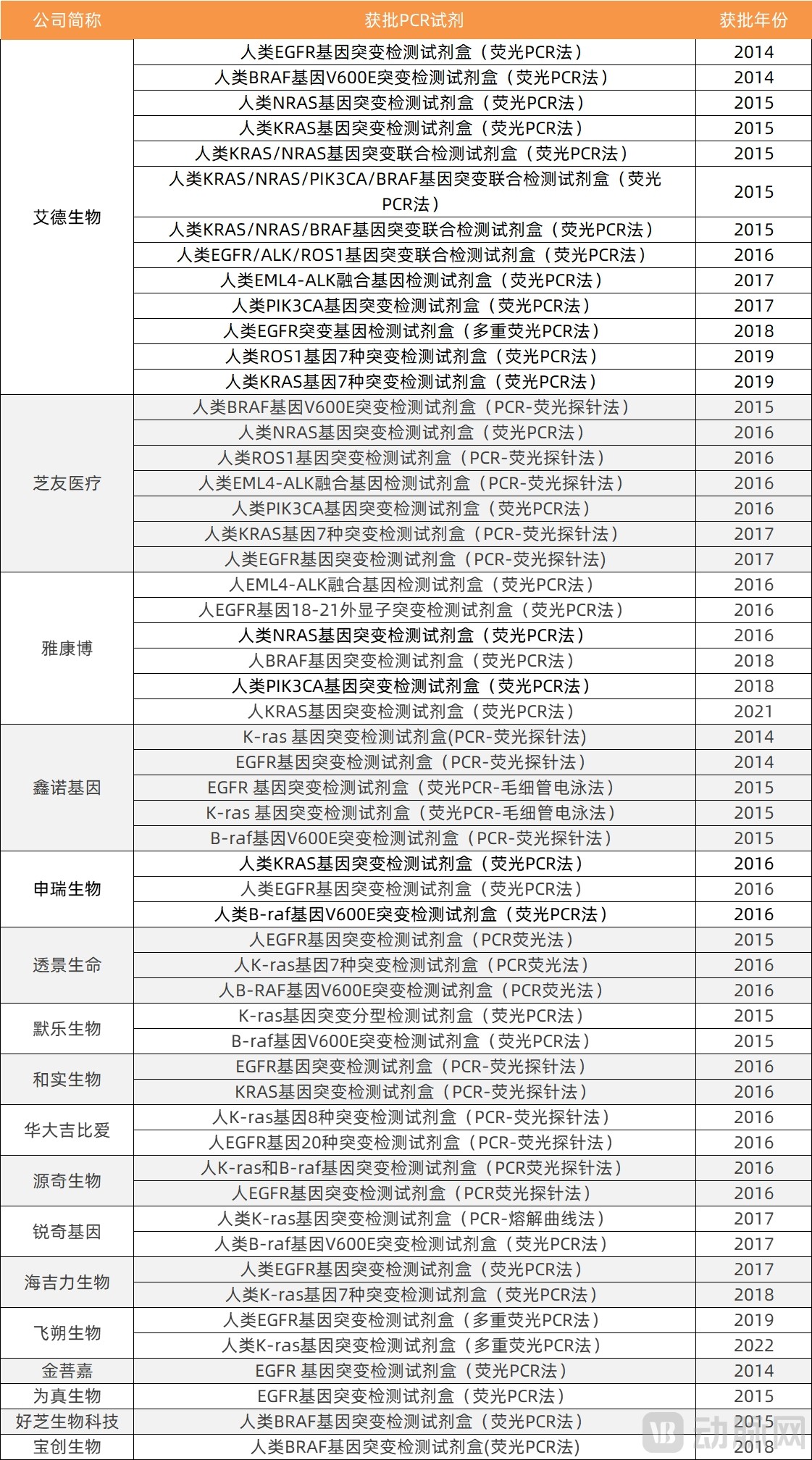
Data Source: National Medical Products Administration
Currently, all NGS products approved for market are small panels; the complexity of clinical trials for large panels has hindered product approval. Specifically, the status of NGS approvals in China is as follows:
NGSStatistics on Approved Reagents for Companion Diagnostics by Technology
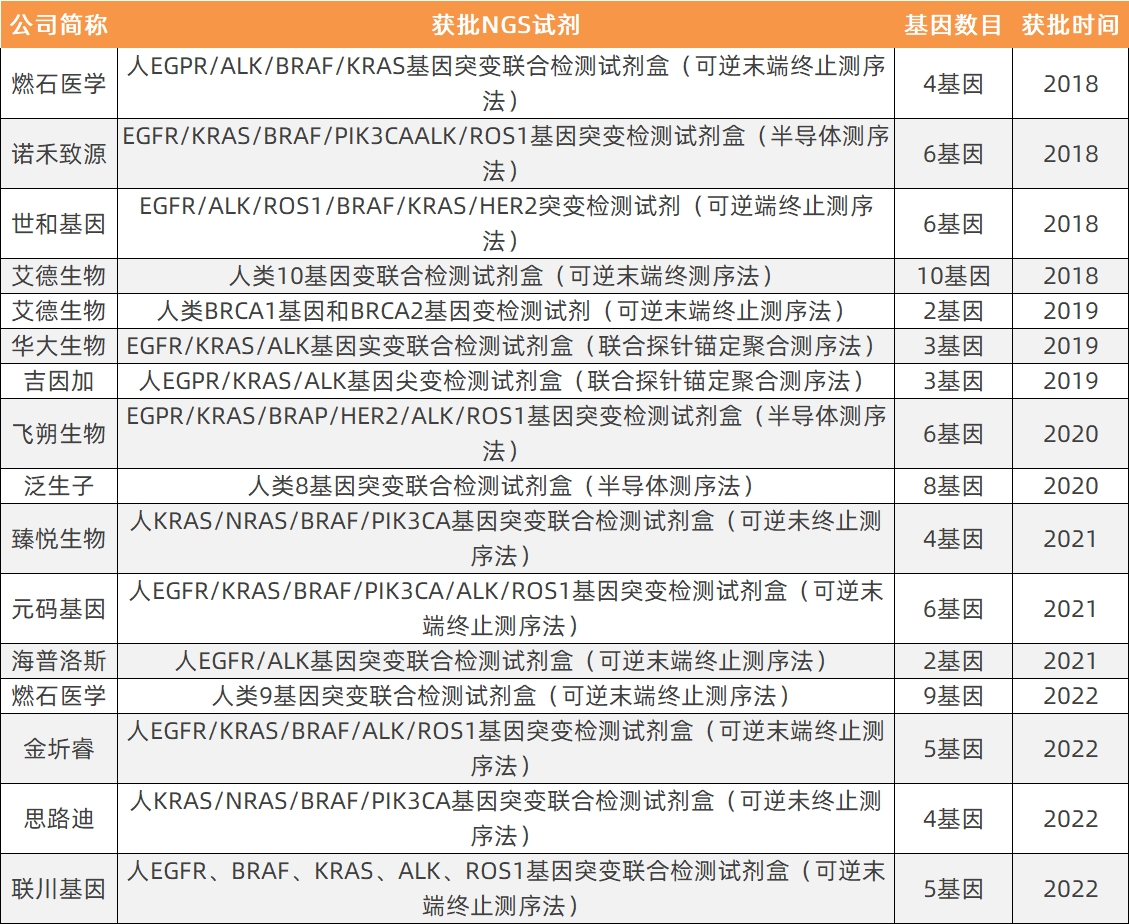
Data Source: National Medical Products Administration
IHC-approved products also entered the market relatively early, with the first approval granted in 2014. Among the approved products, in addition to those used for companion diagnostics, there are also assays for hormone receptor antibody detection. It is evident that, with the rise of immunotherapy in China, two PD-L1 testing kits have already been approved this year.
Statistics on IHC-Based Reagents Approved for Companion Diagnostics
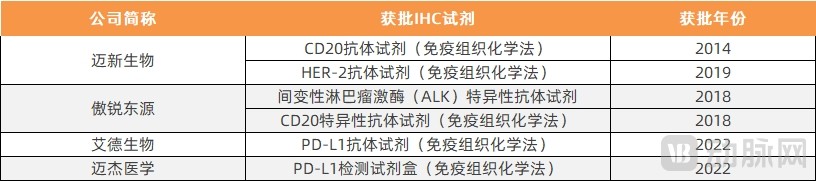
Data Source: National Medical Products Administration
FISH-based products were approved earlier. Among the approved products, in addition to those used for companion diagnostics, there are also reagents used for prenatal diagnosis and other purposes. Among the following reagents, there are specific drugs targeting PML/RARA, BCR/ABL, HER2, and ALK gene mutations, while no specific drugs have been identified for the others.
Statistics on FISH-Based Companion Diagnostic Assays Approved for Use
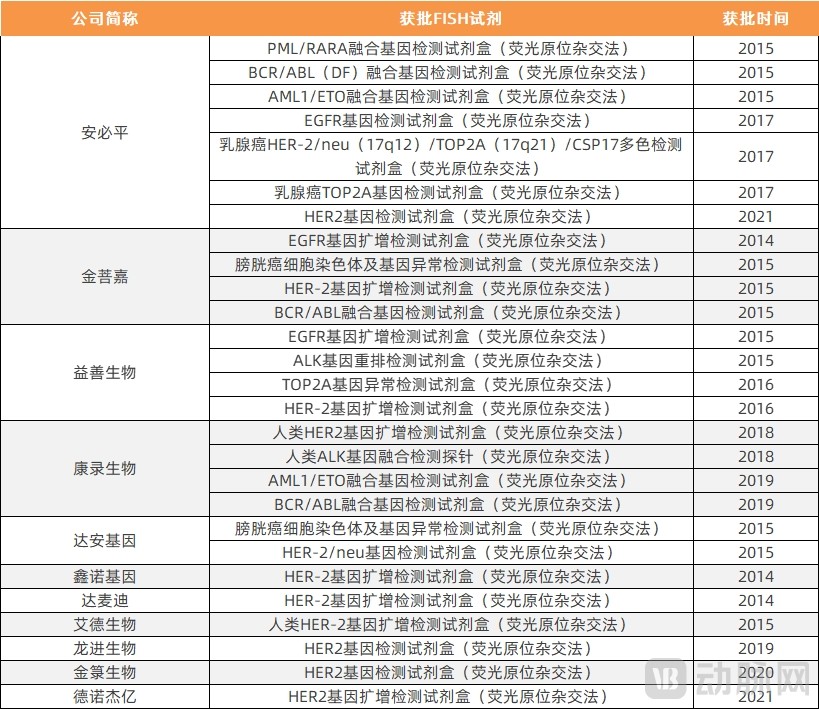
Data Source: National Medical Products Administration
Compared with FDA-approved products in the field of companion diagnostics, there are no approved reagents based on CISH or ISH technologies in China, and few companies are engaged in developing these technologies. Furthermore, the top three FDA-approved technologies are PCR, IHC, and NGS, whereas the top three in China are PCR, FISH, and NGS, with IHC ranking fourth, indicating a significant difference from the United States. The specific number and proportion of approvals by technology are as follows:
Statistical Analysis of the Proportion of Technologies in FDA- and NMPA-Approved Reagents
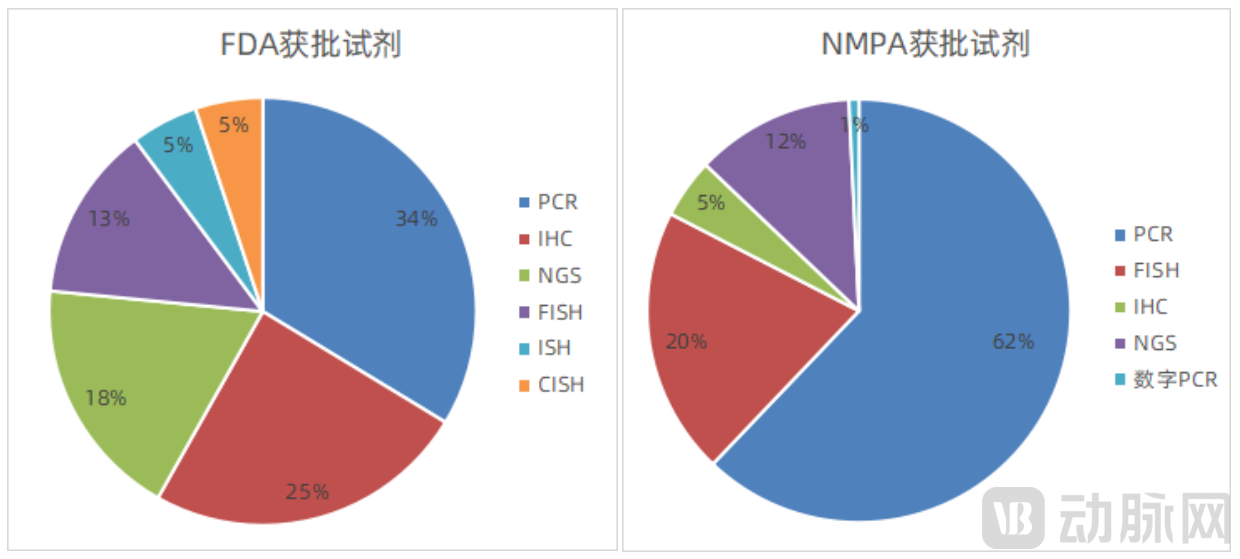
Data Source: National Medical Products Administration
Digital PCR enables direct quantitative analysis.Digital PCR is an emerging nucleic acid detection technology that allocates each nucleic acid molecule to an independent compartment, thereby avoiding interference from selective amplification on amplification results. It enables absolute quantification of nucleic acid templates and facilitates the detection of rare mutations, copy number variations, DNA methylation, and gene rearrangements.
Known as the third-generation PCR, digital PCR enables direct quantitative analysis. It offers advantages such as absolute quantification, high detection sensitivity, the ability to detect low-abundance targets against a high-abundance background, and strong resistance to interference.
Frozen-section immunohistochemistry has expanded the applications of IHC, while multiplex fluorescent immunohistochemistry (mIHC) has upgraded IHC technology.Intraoperative frozen section pathological diagnosis is a commonly used emergency pathological consultation during surgery, serving as a crucial basis for surgeons to formulate surgical plans. The testing time can be reduced from the original 3-4 hours to 15 minutes.
Frozen section immunohistochemistry is primarily used for: (1) determining whether a lesion is neoplastic and assessing its benign or malignant nature; (2) evaluating whether surgical margins are negative; (3) assessing whether the tumor has metastasized to adjacent lymph nodes or organs; and (4) aiding in the identification of certain unexpected or uncertain suspicious minute tissues during surgery.
Multiplex fluorescence immunohistochemistry addresses the limitations of gene expression profiling, flow cytometry, and conventional immunohistochemistry. This technology features three major breakthroughs: it resolves antibody source conflicts during the staining process, enabling simultaneous staining for 7–9 markers; it eliminates spectral crosstalk interference during scanning and imaging, thereby improving the signal-to-noise ratio of images; and it standardizes quantitative analysis in the image analysis phase.
Rapid FISH effectively shortens hybridization time.Currently, the standard hybridization time for FISH probes is 16 hours, with a minimum of no less than 8 hours. The rapid FISH product features the following technical advantages: (1) The required hybridization time can be reduced to 2 hours, significantly shortening the fluorescence in situ hybridization detection time compared to the 16–24 hours required by commercially available FISH probe kits; (2) It significantly reduces non-specific signals, thereby improving the specificity, sensitivity, and accuracy of probe detection.
We analyze the aforementioned technologies based on their maturity, duration of development in China, and current market size in the field of companion diagnostics, as follows:
Analysis of Mainstream Technology Maturity and Market Size
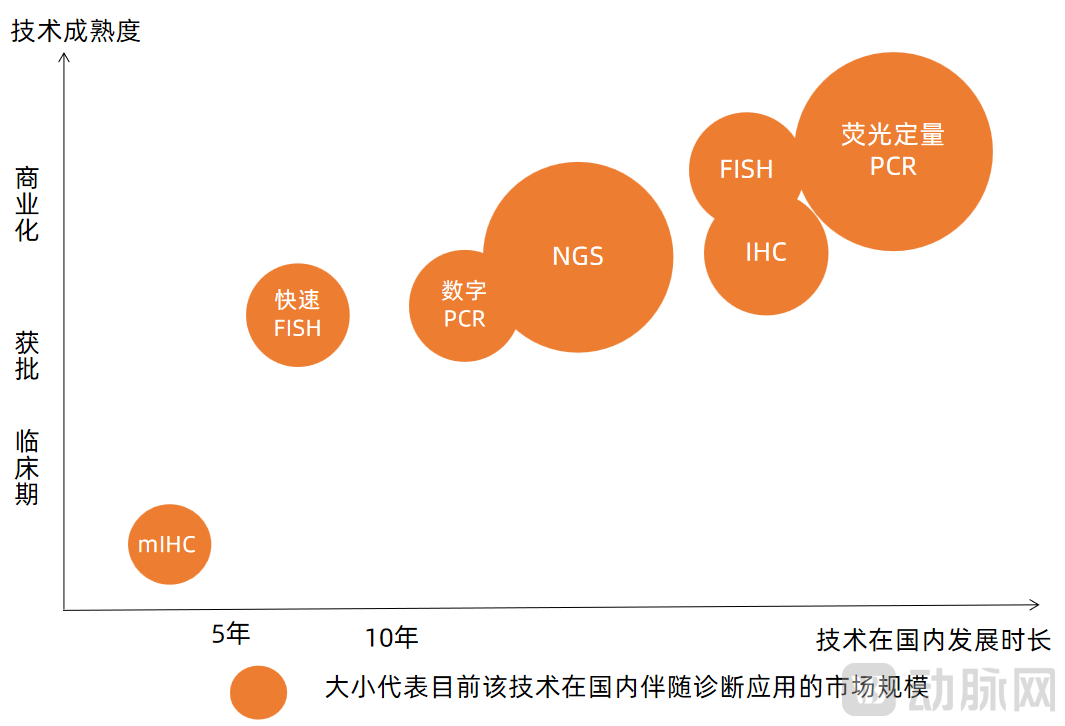
Image source: VCBeat
Emerging Technologies, Multi-Omics Takes Shape
High-throughput single-cell technologies can be categorized into two types: single-cell sequencing for gene-level analysis and mass cytometry (CyTOF) for protein-level analysis. These single-cell technologies enable pharmaceutical companies to conduct high-throughput drug target screening, pharmacokinetic analysis, and efficacy evaluation at the single-cell level, thereby significantly shortening the drug discovery cycle, reducing the costs of new drug development, and optimizing new drug pipelines.
Single-cell sequencing facilitates tumor medication guidance, targeted drug development, and tumor microenvironment analysis.Leveraging single-cell sequencing technology to track cancer cell phenotypes during tumor treatment helps address evolutionary issues such as tumorigenic pathways and drug resistance, thereby significantly influencing future clinical decision-making and the rational design of therapeutic strategies.
Additionally, single-cell sequencing can help identify biomarkers for different types of cancer, providing a critical basis for the diagnosis of cancers of unknown primary origin.
Mass Cytometry AssistantGuidance on oncology medication, development of immunotherapy drugs, and analysis of the tumor microenvironment.In cancer immunotherapy, mass cytometry (CyTOF) characterizes the immune microenvironment features of peripheral blood and tumor tissues in patients treated with immune response-modulating drugs by detecting combinations of multiple markers. This facilitates in-depth analysis of biomarkers, particularly proteins, thereby enabling better assessment of treatment effects on the immune system, identification of prognosis-related biomarkers, and provision of critical guidance for clinical drug administration.
Meanwhile, for key differential cell subpopulations, single-cell sequencing and mass cytometry can be combined to perform joint analysis at the gene and protein levels, thereby enabling proteomic analysis of individual subpopulations, including differential protein analysis, expression pattern analysis, GO enrichment analysis, pathway analysis, and protein–protein interaction analysis.
Joint Analysis of Single-Cell Sequencing and Mass Cytometry
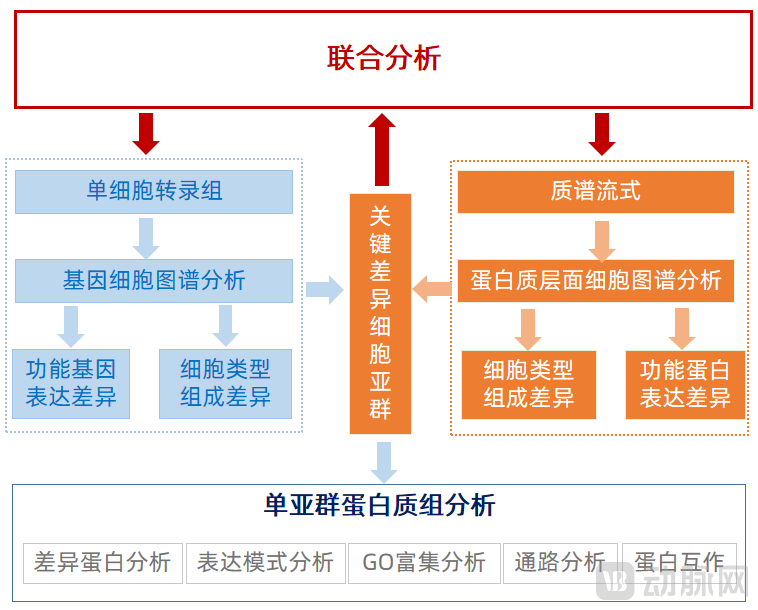
Image source: VCBeat
Mass Spectrometry Facilitates Biomarker Development in Clinical Proteomics. Compared with traditional immunoassays, mass spectrometry enables holistic and comprehensive analysis of diseases at the proteome level, including post-translational modifications of proteins and variants arising from genomic aberrations.
The combined use of mass spectrometry and chromatography can provide important guidance for tumor diagnosis and treatment. In metabolomics research,The combined use of mass spectrometry and chromatography can enhance separation capability and sensitivity. Metabolic changes between tumor cells and normal cells manifest as characteristics such as proliferation, invasion, and metastasis. Additionally, nucleic acid mass spectrometry technology can also facilitate tumor genetic testing.If nucleic acid is usedMass Spectrometry andRT-PCR was used to detect EGFR gene mutations in patients with non-small cell lung cancer. The results from both methods were consistent, and nucleic acid mass spectrometry offers low cost, high throughput, high sensitivity, and full automation.
Multi-omics technologies embody the systems biology approach to biological research.Omics research is a method for investigating the interactions among multiple substances within biological systems, including genomics, epigenomics, transcriptomics, proteomics, metabolomics, and microbiomics. These components collectively influence the phenotypes, traits, and other characteristics of living systems.
As omics research continues to deepen, the integrated analysis of multi-omics data enables a comprehensive and systematic understanding of the interrelationships among various substances in fields such as basic research, clinical diagnosis, and drug development.
Multi-omics technologies can improve the precision of diagnosis and treatment.A common approach to multi-omics integrative analysis involves screening for various target biomolecules, analyzing their functions based on the functional hierarchical logic of systems biology, and performing integrative analysis of transcriptomic, proteomic, and metabolomic data according to co-expression networks and co-regulatory logic.
Through integrated data analysis and mutual validation, we achieve a comprehensive understanding of the major trends and directions in biological changes, propose models of molecular biological change mechanisms, and screen key metabolic pathways, proteins, genes, or metabolites for further in-depth experimental analysis and application. This enables the identification of the most precise biomarkers, thereby facilitating more accurate diagnosis and treatment.
Technical applications must meet the “gold standard”
For a technology to be successfully applied in clinical settings, it must possess certain characteristics. First, it must demonstrate fundamental attributes such as clinical relevance, sensitivity, and stability. Additionally, it must meet further criteria to achieve widespread clinical adoption. Through expert surveys, we selected five indicators: standardization, ease of use, throughput, technological cost, and personnel cost. Among these, standardization refers to the comprehensiveness of regulatory approval frameworks for the technology, while ease of use indicates how straightforward the technology is to operate.
Overall, among mainstream technologies and their upgraded versions, PCR demonstrates the highest level of standardization and ease of use, NGS offers the greatest throughput, and IHC incurs the lowest cost. Emerging technologies generally exhibit lower levels of standardization and higher costs, remaining some distance away from successful clinical application—a characteristic commonly observed in the adoption of most emerging technologies. The specific ratings are as follows:
Analysis of Mainstream Technologies and Their Upgraded Technical Features
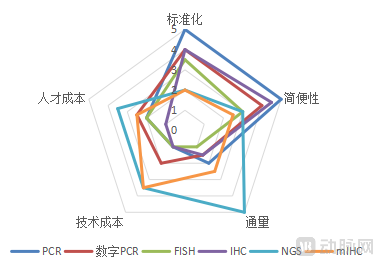
Image source: VCBeat
Analysis of Emerging Technology Characteristics
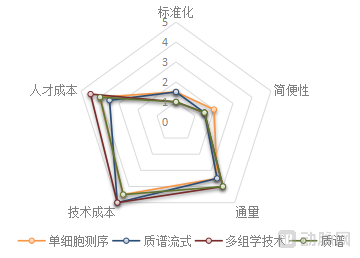
Image source: VCBeat
AI-Empowered Pathological Interpretation and Bioinformatics Analysis
AI-driven pathological interpretation offers advantages in speed, accuracy, and multi-dimensional analysis. Leveraging AI-based bioinformatics analysis to support the development of companion diagnostic products represents a future direction. On one hand, unlike traditional medical devices, AI-enabled medical devices can continuously learn and optimize in real-world settings, leading to ongoing performance improvements; furthermore, real-world studies can provide supporting evidence at the time of drug market approval.
On the other hand, the human genome comprises more than 20,000 genes. Leveraging AI technologies to conduct integrated multi-omics analyses of genomic, proteomic, and other data enables precise matching of patients with appropriate companion diagnostic products. This represents a long-term strategic vision for AI applications in companion diagnostics.
It’s the Right Time to Go Global: Co-Development with Pharmaceutical Companies Is Gaining Momentum
Mainstream: Complementary coexistence of in-hospital and out-of-hospital models, with inconsistent regulation
There are two models for the clinical adoption of companion diagnostic products: the in-hospital model and the out-of-hospital model. Generally, the distinction between the two lies in whether the product has received regulatory approval. Approved products can be introduced into hospitals as in vitro diagnostic (IVD) reagents, whereas unapproved products can only be offered as laboratory-developed test (LDT) services outside of hospitals. The specific models are as follows:
Characteristics of the In-Hospital and Out-of-Hospital Model
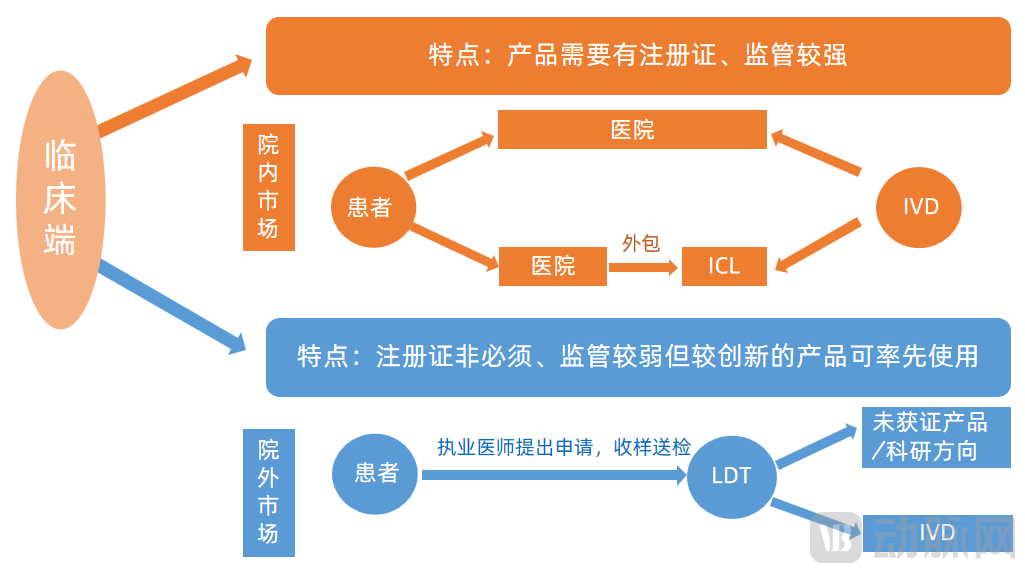
Image source: Burning Rock Biotech, Genetron Health prospectus
China’s LDTs See First-Ever Regulations; In-House LDTs Gradually Being Implemented2021March 18, GuoThe National Medical Products Administration (NMPA) website released the revised "Regulations on the Supervision and Administration of Medical Devices," in which Article 53 provides clear regulatory requirements for the use scenarios of Laboratory Developed Tests (LDTs). This marks the first time that policies have explicitly set forth requirements regarding LDT use scenarios and standards.
On July 15, 2021, Article 4 of the “Opinions of the Central Committee of the Communist Party of China and the State Council on Supporting Pudong New Area in Carrying Out High-Level Reform and Opening-Up and Building a Leading Zone for Socialist Modernization” explicitly stated: Within the jurisdiction of Pudong New Area, qualified medical institutions are permitted to conduct pilot programs for the in-house development of in vitro diagnostic reagents in accordance with relevant requirements.
On October 9, 2022, the Office of the Leading Group for Deepening the Reform of the Medical and Healthcare System in Shanghai issued the “Notice on Launching Pilot Work for the High-Quality Development of Public Hospitals in Shanghai.” Relevant departments selected 40 public medical institutions as pilot units and designated 20 public hospitals as guided pilot units for reference implementation, with the pilot program spanning five years. This notice advanced the implementation of the Laboratory Developed Tests (LDT) model in hospital pilots. Although it did not specify which hospitals were permitted to conduct LDT pilots, this marked the first time that participating institutions were selected and their names publicly disclosed. The industry generally believes that public Grade A tertiary hospitals are more likely to be chosen for LDT pilots.
On one hand, launching LDT pilots in public tertiary hospitals makes the regulation of LDTs more manageable; on the other hand, while LDT products inherently carry risks, tertiary hospitals possess a stronger capacity to assume such risks. Although no more stringent regulatory policies for LDTs have been introduced to date, enterprises whose LDT products gain access to tertiary hospitals inevitably demonstrate superior R&D capabilities and comprehensive strength.
In other words, gaining access to pilot hospitals is itself a threshold; as such, LDT regulation may be entering a new phase.
Channels: Overseas expansion has seen significant growth in recent years, while penetration into lower-tier markets remains in its early stages.
In recent years, with the outbreak of the COVID-19 pandemic, in vitro diagnostic (IVD) reagents, as representative medical devices for domestic substitution, have seen a gradual increase in their share of exports, continuously optimizing the category structure of medical device exports. Their export share has grown from 1.3% in 2019 to 23% in the first half of 2022, showing the most significant growth trend.
Export Performance of Various Categories of Medical Devices in China (First Half of 2019–2022)
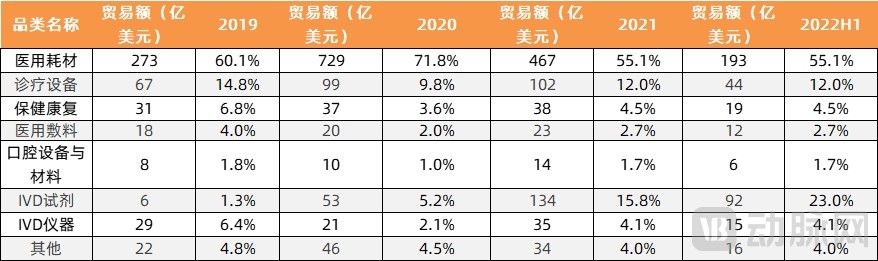
Image source: VCBeat
In terms of overseas expansion destinations, the primary factors considered by enterprises are registration difficulty and product maturity, with a positive correlation between the two. Regarding the markets in the United States, Japan, Europe, the Middle East, Southeast Asia, and Africa, our analysis from the perspectives of product maturity and registration difficulty is as follows:
Distribution of Registration Difficulty and Maturity for Companion Diagnostic Products Across Regional Markets
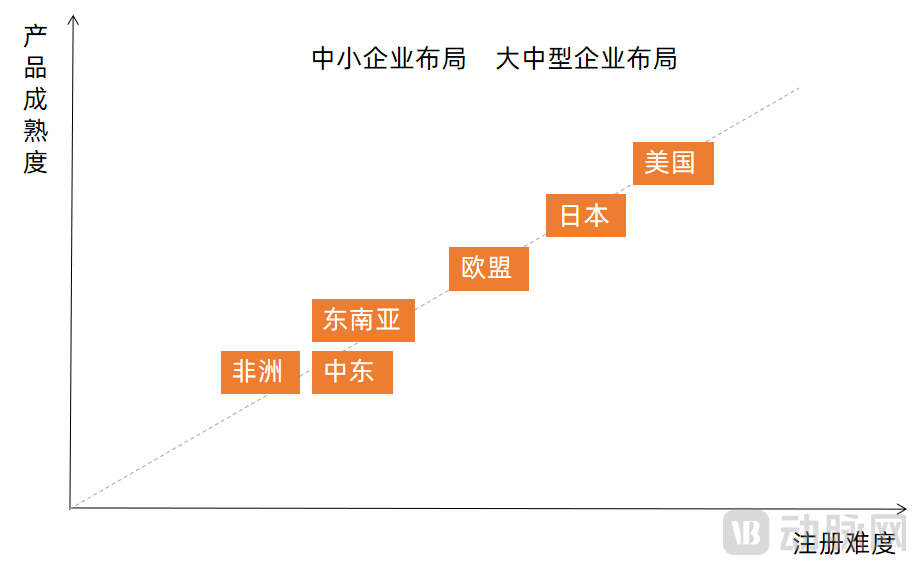
Image source: VCBeat
Going global primarily faces two major challenges: regulatory compliance and partner selection.Regulatory compliance is the first priority. To achieve long-term development overseas, product registration is mandatory. Given the varying policy environments and regulatory intensities across different countries, enterprises must have a thorough understanding of overseas registration regulations. While the registration process is similar to that in China, there are differences in specific details, such as whether manufacturing can take place in China and whether the company itself must act as the registrant.
Securing win-win, long-term partners is half the battle in successful global expansion. There are generally three strategies for going global: distributor model, subsidiary plus distributor model, and wholly-owned subsidiary model. Overall, the overseas expansion of Chinese enterprises has surged in recent years due to the COVID-19 pandemic and remains in its early stages, with the distributor model being the predominant approach.
Overseas partners generally include distributors, agents, pharmaceutical companies, and regulatory registration partners. Collaborating with multinational pharmaceutical companies to co-develop companion diagnostic products represents the optimal strategy, although this approach demands substantial corporate capabilities. When selecting distributors and agents, emphasis should be placed on their channel capabilities. Partners can help enterprises address challenges related to distribution channels and regulatory registration; therefore, identifying partners who foster win-win, sustainable collaborations is crucial.
Leveraging both internal and external forces is the sustainable path for global expansion.Turning to the companies themselves, what commonalities do successful globalizing enterprises share? According to the Globalization Index released by VCBeat, a company’s capability for international expansion is assessed across five key dimensions: overseas registration, patents and software copyrights, clinical capabilities, R&D strength, and revenue performance. These represent the core internal competencies that companies must cultivate.
Meanwhile, we can anticipate that as companies continue to strengthen their internal capabilities, more will embark on overseas mergers and acquisitions (M&A) or leverage capital to expand into global markets. For instance, companies may target firms with strong technological advantages, distribution channels, or both. Given the high concentration of technology-driven enterprises in European and American markets, these regions represent optimal targets for outbound M&A.
Of course, it must be noted that overseas regulations are becoming increasingly stringent. For instance, the European Union’s new In Vitro Diagnostic Regulation (IVDR) came into effect on May 26, 2022. The IVDR reclassifies in vitro diagnostic products, significantly increasing the difficulty of market entry after its implementation.
Equipment, pharmaceuticals, and physicians have become the key barriers to penetrating grassroots-level healthcare.Supply and demand are difficult to balance.From the demand side, there is a large and dispersed population of cancer patients at the primary care level, coupled with low awareness of cancer prevention. From the supply side, first, the distribution of oncologists is uneven, creating a strong siphon effect; second, equipment and medications pose significant challenges. Cancer diagnosis and treatment require sophisticated medical instruments. The high capital investment needed for follow-up examination equipment and chemotherapy devices deters secondary hospitals, let alone ensuring the availability of targeted therapies at the primary care level.
As companion diagnostic products trickle down from tertiary hospitals to secondary hospitals, we believe this process can be divided into three stages, primarily characterized by varying degrees of adoption in equipment, drugs, and physician expertise.
Three Stages of Companion Diagnostics Penetrating into Primary Care
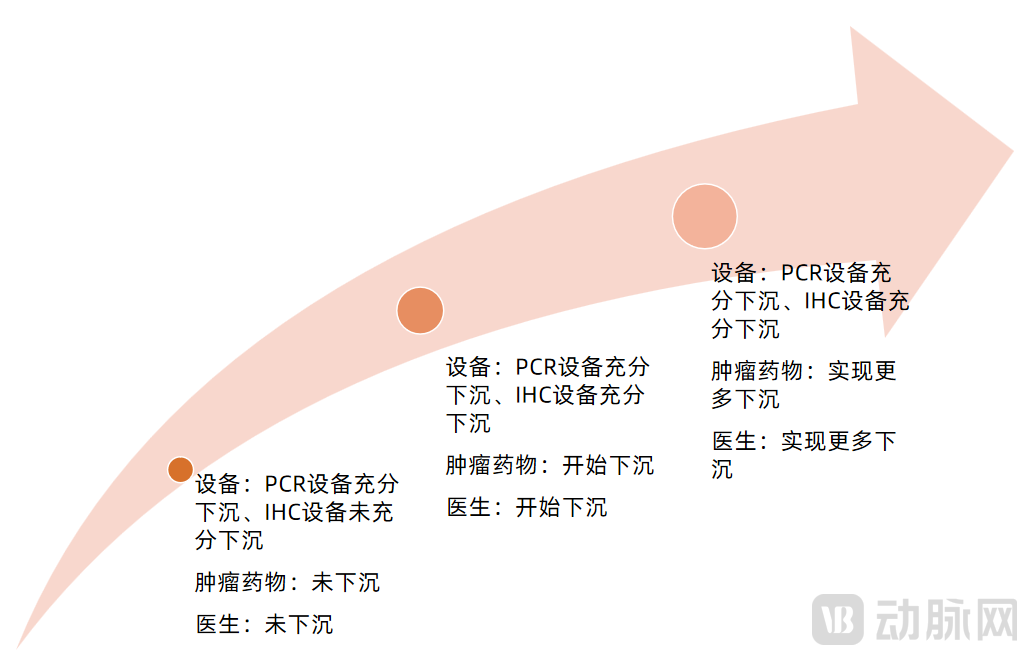
Image source: VCBeat
Extension: Co-establishing in-house laboratories and joint development with pharmaceutical companies
Overall, hospital-based co-built laboratories primarily focus on the clinical application of innovative technologies, aiming to advance and promote their adoption. They serve as a complement to third-party medical laboratories and represent a transitional form from the Laboratory Developed Tests (LDT) model to the In Vitro Diagnostics (IVD) model.
Among the technologies applied to companion diagnostics, NGS-based co-established laboratories are currently the mainstream approach. The participating companies are predominantly industry leaders, and the relationship between enterprises and hospitals is not one-to-one but rather many-to-many.
Co-built laboratories within hospitals are a double-edged sword for enterprises.The joint laboratory model addresses the challenges of implementing new technologies in hospitals by assisting them in establishing standardized testing laboratories through support in both hardware (equipment) and software (technical training and quality control). This enables rapid, accurate, and compliant genetic testing, benefiting hospitals while facilitating market access for enterprise products and securing a first-mover advantage.
However, current practices indicate that due to the high costs associated with co-building initiatives involving next-generation sequencing (NGS), most enterprises remain in a loss-making position in the short term.
In the long run, as physicians at large tertiary hospitals become more familiar with NGS applications through corporate training, the barriers to establishing in-house hospital laboratories will diminish, thereby reducing their dependence on enterprises. Meanwhile, products entering hospitals are highly susceptible to policy influences; once NGS products mature, the prevailing environment of centralized procurement and cost containment may lead to a decoupling of enterprise products from hospitals.
For secondary hospitals, at the current stage, co-built laboratories are more suitable for routine testing projects, given the high costs of next-generation sequencing (NGS) and the challenges in decentralizing tumor companion diagnostic products. Overall, co-built laboratories are better suited for the initial implementation of capital-intensive new technologies at top-tier tertiary hospitals, led by industry-leading enterprises. On the other hand, they also facilitate the adoption of less costly technologies in secondary hospitals.
On June 28, 2022, the “Guiding Principles for Clinical Trial Registration and Review of Original Companion Diagnostic Reagents Developed Concurrently with Antineoplastic Drugs” was officially released. The guidelines aim to promote the concurrent development of pharmaceuticals and companion diagnostic products, encouraging collaboration between pharmaceutical companies and companion diagnostic developers from earlier clinical stages. There are three main development models for companion diagnostic products: concurrent, bridging, and follow-on.
Three Models for the Development of Companion Diagnostic Products
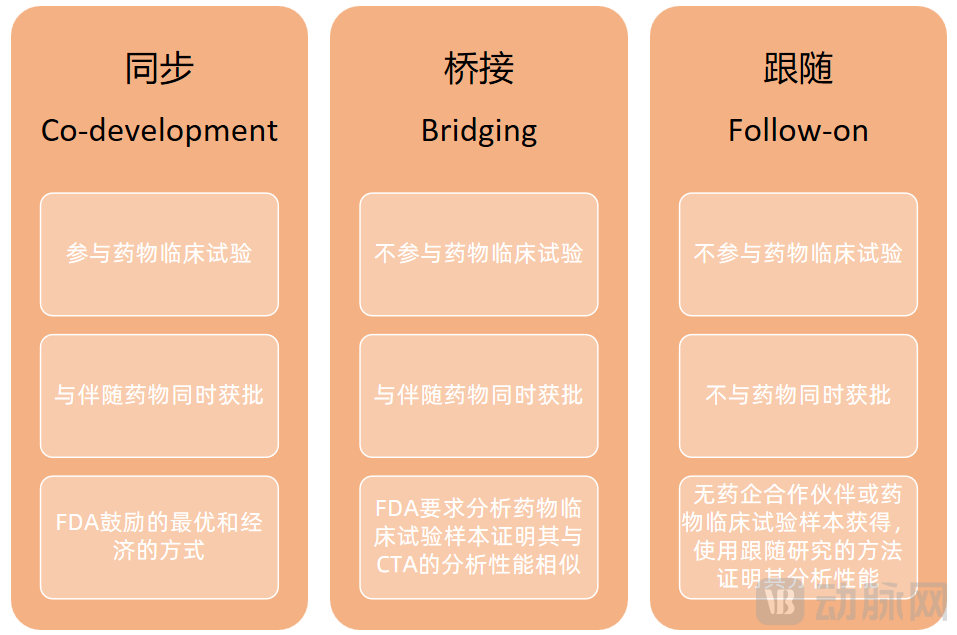
Image source: VCBeat
More innovative targets are the focus of collaboration between pharmaceutical companies and companion diagnostic firms.There are numerous potential drug targets, and the corresponding companion diagnostic kits are equally diverse and complex. Each drug targeting a specific marker must be used in conjunction with its own dedicated companion diagnostic kit to truly achieve precision therapy.
Some well-developed biomarkers, such as EGFR, ALK, and PD-L1, already have multiple companion diagnostic products on the market, with the competitive landscape shifting from a blue ocean to a red ocean. In contrast, more innovative targets remain entirely in a blue ocean state and represent the key focus of collaboration between pharmaceutical companies and companion diagnostic firms.
Collaborations between pharmaceutical companies and companion diagnostic providers are progressively shifting earlier along the drug development timeline.In fact, many drugs in the clinical stage have begun to develop supporting companion diagnostic products and incorporate them into clinical trials simultaneously. Integrating companion diagnostics with new drug development not only optimizes the efficiency of companion diagnostic reagent development but also reduces the risks associated with new drug clinical trials to a certain extent.
In new drug clinical trials, companion diagnostics can precisely identify target populations and indications by stratifying or enriching patient cohorts, thereby helping achieve more favorable clinical outcomes, shortening the drug development cycle, controlling trial scale, and improving the success rate of clinical trials.
The early involvement of companion diagnostics also brings greater challenges.In the intricate landscape of new drug development, simultaneously optimizing companion diagnostic product development and the efficiency of clinical trials for novel therapeutics is no easy feat. There are three key considerations.
First, it is necessary to select companion diagnostic products with sufficiently high sensitivity and specificity. For the development of companion diagnostic kits, performance validation is a core component, requiring comprehensive consideration of four aspects: accuracy, limit of detection, precision, and specificity.
Second, companies developing companion diagnostics must possess considerable technical reliability and stability. Over the past decade, companion diagnostic technologies have diverged significantly. In biomarker selection, there has been a shift from single targets to multi-target panels; sample types have expanded from predominantly tissue-based samples to diverse options including liquid biopsy specimens; and detection technologies have continuously evolved, transitioning from previously dominant methods such as qPCR, IHC, and FISH targeting single markers to various NGS panels characterized by biomarker enrichment.
Third, diagnostic companies developing companion diagnostics must possess extensive regulatory experience and a robust regulatory affairs team. Incorporating companion diagnostic development into new drug clinical trials requires close collaboration and joint efforts between pharmaceutical companies and diagnostic firms. Both parties’ teams are responsible for securing regulatory approval for their respective products, ensuring that the market authorization process is completed synchronously and as early as possible.
Overall, the companion diagnostics industry for precision-matched drugs is emerging in China.This trend is a cross-product of the booming domestic innovative drug industry and genetic testing sector, and it also reflects the broader momentum toward precision medicine.
LDTs Prioritize Implementation in Tertiary Grade A Hospitals, with Expanded Insurance Coverage for Companion Diagnostics
Technology: Mainstream and Emerging Technologies Drive Mutual Advancement
PCR and gene sequencing have been upgraded to third-generation technologies; mIHC enables multiplex labeling, and FISH is faster.A Review of Mainstream Technologies Applied in Companion Diagnostics in Recent Years: Both PCR and gene sequencing have advanced to third-generation platforms. Immunohistochemistry (IHC) has seen the emergence of intraoperative frozen section IHC solutions, as well as breakthroughs in multiplex IHC (mIHC), evolving from single-marker detection to simultaneous labeling of 7–9 biomarkers. Traditional FISH technology, characterized by long hybridization times and slow turnaround, has addressed this bottleneck with the development of rapid FISH assays.
Single-Cell Technologies and Mass Spectrometry Inject New Vitality into Companion Diagnostics.With breakthroughs in core technologies, new technologies have gradually come into the public eye in recent years, with emerging approaches such as single-cell analysis and mass spectrometry garnering significant attention.
Single-cell technologies interpret and analyze changes at the cellular level. Single-cell sequencing for genomic research and mass cytometry for proteomic research represent the core directions of high-throughput single-cell technologies, enabling integrated analysis of genes and proteins. Meanwhile, mass spectrometry has emerged as the most important analytical technique in the fields of protein research and biomacromolecule studies, garnering significant attention in recent years.
Multi-technology and multi-omics approaches will jointly drive industry development.On one hand, technologies vary in their characteristics, complement and synergize with each other, and co-develop; meanwhile, the closed-loop application of AI will empower these technologies.
On the other hand, a systems-thinking approach to multi-omics may propel biology to new heights. Currently, integrated, multi-dimensional analyses across genomics, proteomics, metabolomics, and other omics disciplines are poised to deliver more precise insights. In the future, the convergence of diverse technologies and multi-omics platforms will jointly drive the development of the companion diagnostics industry, facilitating its advancement toward personalized medicine.
Regulation: Hospital Adoption of LDTs as an Entry Barrier, with Varying Levels of Recognition
In China, thresholds may be established by requiring laboratory-developed tests (LDTs) to be introduced into hospitals rather than through an approved regulatory pathway.LDTs facilitate the commercialization of innovative products, yet they are also prone to market heterogeneity and inconsistent product quality. While the United States has established approval pathways for NGS-based LDTs, China currently lacks such mechanisms. Will China introduce corresponding approval pathways for LDTs to strengthen regulatory oversight?
We believe that, against the backdrop of the ongoing implementation of Laboratory Developed Tests (LDTs) pilot programs in hospitals this year, China may establish entry barriers by requiring LDTs to be adopted directly within hospitals, rather than by creating a formal approval pathway.
Hospital adoption of LDTs serves as the entry threshold, with varying levels of recognition across different collaborations.Whether through co-established laboratories or collaborative models with pilot hospitals, barriers remain for the adoption of Laboratory Developed Tests (LDTs) in healthcare institutions. Under these barriers, product acceptance inevitably varies; for instance, due to differences among pilot hospitals, the level of recognition for pilot LDT products differs, thereby affecting the difficulty of product registration.
In the future, we believe that it will be less difficult to obtain approval for LDT products piloted in large tertiary Grade A hospitals than for those piloted in secondary hospitals or third-party testing institutions.
Competition among enterprises may center on hospitals collaborating on LDT products.Where LDT products are developed will become a new benchmark, and competition for channels among pilot tertiary Grade A hospitals will intensify. Only enterprises with both strong R&D and marketing capabilities will prevail. In addition to public tertiary Grade A hospitals, secondary hospitals and third-party testing laboratories may also be included in the LDT pilot programs.
However, in terms of regulatory complexity, we believe that LDT pilots at this stage are more likely to be implemented in large public tertiary hospitals, while it is unlikely for secondary hospitals or third-party testing institutions to become pilot sites in the short term.
Payment: Medical insurance accelerates expansion; PCR may be the first to undergo centralized procurement
The inclusion of tumor genetic testing services in the national health insurance scheme will be further accelerated.Regarding payment, currently only Beijing has included companion diagnostic products in its medical insurance coverage. Commercial insurance plans mostly cover these as technical procedures, such as NGS testing items, rather than as companion diagnostic items.
Companion diagnostics are an essential requirement. It is reported that the National Reimbursement Drug List includes more than 30 targeted therapies, and according to the relevant requirements for restricted reimbursement coverage, genetic testing is required for the use of specific targeted drugs.
On October 12, 2022, the National Healthcare Security Administration (NHSA) issued an official response regarding the inclusion of oncology genetic testing services in the national medical insurance scheme and their participation in volume-based procurement: certain regions have already incorporated selected genetic testing items into the scope of medical insurance coverage. Furthermore, genetic testing services that are “safe and effective, cost-appropriate, and have clearly defined charging standards” are also expected to be included in the medical insurance coverage.
In the short term, NGS is unlikely to achieve comprehensive coverage.In the long term, including tumor genetic testing in medical insurance will facilitate the implementation of companion diagnostic projects in hospitals; however, in the short term, NGS is unlikely to be covered. Technically, PCR and IHC are currently included in the national medical insurance catalog, while NGS is not. As is well known, NGS products are expensive, with prices ranging from several thousand to over ten thousand yuan.
However, NGS requires substantial upfront investment and a lengthy approval process, typically taking 3–5 years. Large-panel products are still under development. If included in the National Reimbursement Drug List (NRDL) at this stage, companies’ profit margins would shrink, jeopardizing their R&D investments. Therefore, we believe that NGS will not be included in the NRDL in the short term; reimbursement may be deferred until costs decline further.
Centralized Procurement Is Inevitable; PCR May Bear the Brunt.Amid the broader context of centralized procurement, will oncology genetic testing be subject to volume-based procurement? The National Healthcare Security Administration further stated, “Oncology genetic testing is classified as a medical service item. Under current policies, basic medical services provided by public healthcare institutions are subject to government-guided pricing. Currently, we are guiding local authorities to explore centralized procurement of diagnostic reagents in accordance with the principle of ‘separating technical service fees from consumable costs,’ thereby promoting the return of reagent prices to reasonable levels and driving down the prices of related medical service items.”
On the other hand, in September this year, the National Healthcare Security Administration (NHSA) explicitly stated for the first time that innovative medical devices would not be temporarily included in centralized volume-based procurement (VBP). The NHSA pointed out that “due to the immaturity of clinical application and the difficulty in temporarily estimating the usage volume of innovative medical devices, it is currently challenging to implement a volume-based approach. During the centralized VBP process, the NHSA will reasonably determine the volume-based procurement ratio based on factors such as clinical usage characteristics, market competition landscape, and the number of selected enterprises, thereby reserving a certain portion of the market outside of centralized VBP to create space for the market development of innovative products.”
As such, PCR may be the first to undergo centralized procurement. Currently, PCR is covered by medical insurance and meets the criteria of wide application, mature development, and low cost, with PCR equipment already deployed at the primary care level. Therefore, PCR is the optimal candidate for centralized procurement.
The above is an excerpt of the main content of the report. The complete framework of the report is as follows:
Chapter 1Overview:Policy Gradually Becomes More Standardized, a blue-ocean market worth over RMB 10 billion
1.1Policy: China Enters an Adjustment Phase, with Continuous Standardization
1.2Financing: Recent3Year53Jiahuorong, Latest FinancingBMost Wheels
1.3Market: Late Start, Rapid Growth, Huge Potential
Chapter 2 Technology: Upgrades to Four Mainstream Technologies and Accelerated Adoption of Three Emerging Technologies
2.1Mainstream technologies have been upgraded,PCRandNGSIt is the main force.
2.2The Dawn of Multi-Omics Amidst the Rise of New Technologies
2.3 AIEmpowering Pathological Interpretation and Bioinformatics Analysis
Chapter 3 Commercialization: The Time Is Ripe for Global Expansion, and Co-Development with Pharmaceutical Companies Is Gaining Momentum
3.1Mainstream:Complementary Coexistence of In-Hospital and Out-of-Hospital Models with Divergent Regulatory Oversight
3.2Channel: Overseas expansion has shown significant growth in recent years, while penetration into lower-tier markets remains in its early stages.
3.3Extension: Co-established in-hospital laboratories, joint development with pharmaceutical companies
Chapter 4 Trend: LDTs to be prioritized in tertiary hospitals; companion diagnostics included in expanded medical insurance coverage
4.1Technology: Mainstream and Emerging Technologies Drive Mutual Progress
4.2Regulation:LDTHospital Access as a Threshold, with Varying Levels of Acceptance
4.3Payment:Medical InsuranceAccelerated Expansion,PCRor be the first to undergo centralized procurement
Chapter 5 Corporate Case Studies
5.1 Genetron Health—Building Comprehensive Companion Diagnostic Solutions
5.2 Huisuan Gene – LayoutAI+Precision Diagnosis and Treatment: Delivering Comprehensive Solutions
5.3 Putian Bio—Genetics-Protein-Metabolic "Trinity" Multi-Omics Analysis
5.4 Kuoran Bio—Strategic LayoutNGP+NGS, providing one-stop solutions
5.5 Kanglu Bio—FISHandPCRJoining Forces to Deliver Precision Diagnostics
5.6 Aimu'en—Providing Immune-Driven Medical Solutions
5.7 Sinotest Biotech—Deeply Committed to Oncology Diagnostics, Providing Comprehensive Pathology Solutions
Scan the QR code below to get the full report for free.

Special Thanks(In the order of research interviews):
Mr. Marco, Managing Director of Qiao Capital; Dr. Zhang Yafei, Chairman and CEO of MJ Biomedical; Ms. Xia Lin, CEO of HuiSuan Genomics; Mr. Sun Tao, CEO of Aimune; Ms. Sun Lin, Brand Director of Putian Biology; Aden Biopharmaceuticals; Mr. Sun Hua, Senior Vice President of Kanglu Biotechnology; Mr. Bu Lingbin, CEO of Kuoran Biology; ChenAn Bio; and Mr. Qi Hua, Founder and CEO of SinoCellTech.