Deep Potential Unveils RiDymo™ Platform Pipeline Advancement in Ferroptosis-Targeted Drug Discovery
Ferroptosis, a form of iron-dependent programmed cell death discovered in 2012 by Brent R. Stockwell’s laboratory, is triggered by uncontrolled lipid peroxidation and subsequent plasma membrane rupture, and is closely associated with tumorigenesis, progression, and drug resistance. As a novel mode of cell death distinct from apoptosis, necrosis, and autophagy, ferroptosis has rapidly sparked a global surge in research since its inception.[1]。
Current research indicates that loss of function of GPX4 (glutathione peroxidase 4), a core regulator of ferroptosis, in non-small cell lung cancer, pancreatic cancer, prostate cancer, and melanoma cells can induce sustained ferroptosis in cancer cells, thereby inhibiting tumor initiation, progression, recurrence, metastasis, and drug resistance. Consequently, GPX4 has gradually emerged as a “star target” in the field of ferroptosis research.[2]。
Stuart L. Schreiber, a professor and co-founder of the Broad Institute of MIT and Harvard, has dedicated ten years to this field and founded Kojin Therapeutics. On June 9, 2021, the company announced the completion of a $60 million Series A financing round to accelerate the development of drugs targeting drug-resistant tumors that are sensitive to ferroptosis.
Although GPX4 appears promising, the entire protein is small with a flat surface and lacks druggable pockets (Figure 1A). Currently, GPX4 inhibitors with cellular activity primarily form covalent bonds with the selenocysteine residue at position 46, leading to widespread issues of poor selectivity and high toxicity (Figure 1B).
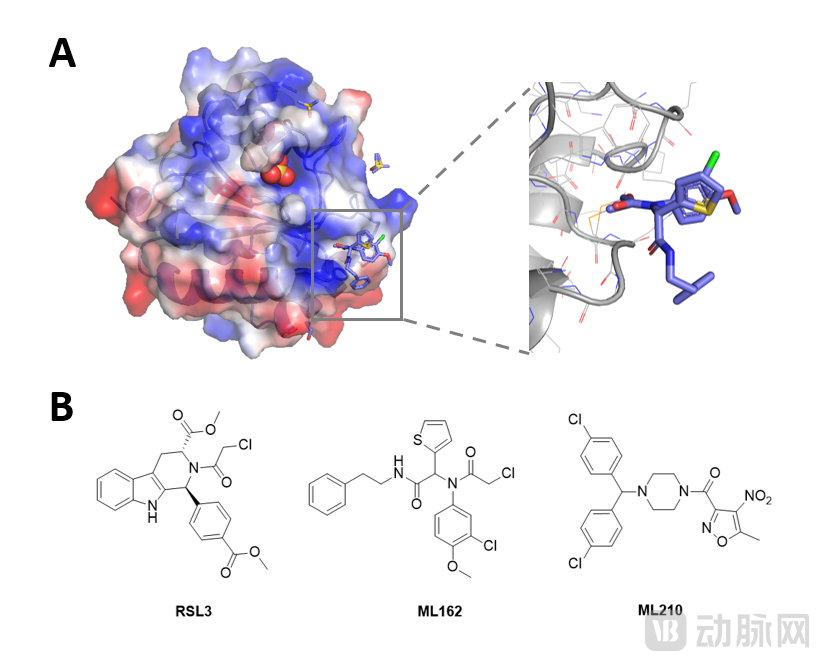
Figure 1. Structure of the GPX4 protein–ML162 crystal complex[3](PDB ID: 6HKQ) and existing covalent inhibitors
A close examination of the interaction between ML162 and GPX4, as shown in Figure 1A, reveals that due to the flat protein surface, there are very few non-covalent interactions between ML162 and GPX4, which may also be the reason for the poor selectivity of the ML162 molecule.
Based on the above, it is highly worthwhile to explore whether GPX4 possesses a hidden pocket or an allosteric pocket, and whether targeting alternative sites can modulate the biological function of GPX4.
“From the outset, our strategy for GPX4 development has been focused on exploring and identifying hidden pockets with superior druggability,” said Dr. Zhang Xiaomin, Chief Pharmaceutical Officer at DP Technology. “Leveraging the RiDymo platform, built on AI for Science, DP Technology utilizes molecular dynamics simulations to uncover conformational changes of proteins in vivo, thereby revealing novel druggable pockets. The non-covalent molecules developed based on these findings demonstrated inhibitory activities comparable to those of positive control covalent molecules at both enzymatic and cellular levels, while exhibiting improved druggability. This practice has fully validated the value of the RiDymo platform in drug R&D, holding promise for empowering drug discovery against more challenging, hard-to-drug targets in the future.”
In this project, the development path of DP Technology’s drug R&D team is as follows:
1. Protein Conformational Sampling: Computational simulations of the GPX4 protein were performed using the Reinforced Dynamics (RiD) method. As shown in the right column of Figure 2, compared with conventional Molecular Dynamics (MD) simulations (left column of Figure 2), RiD can more efficiently explore the protein’s phase space within the same simulation timescale, thereby identifying other metastable conformations and hidden sites (the pink regions indicate amino acids buried within the protein interior).
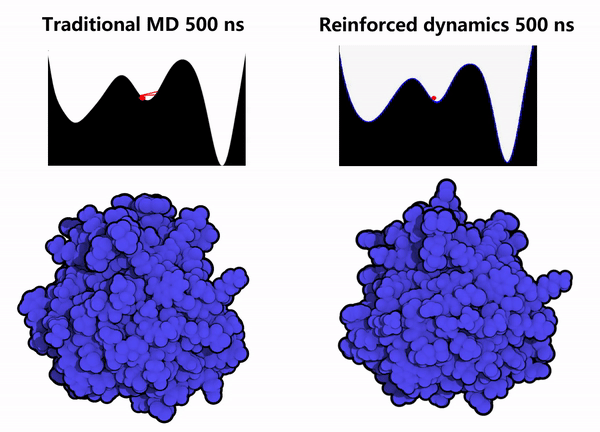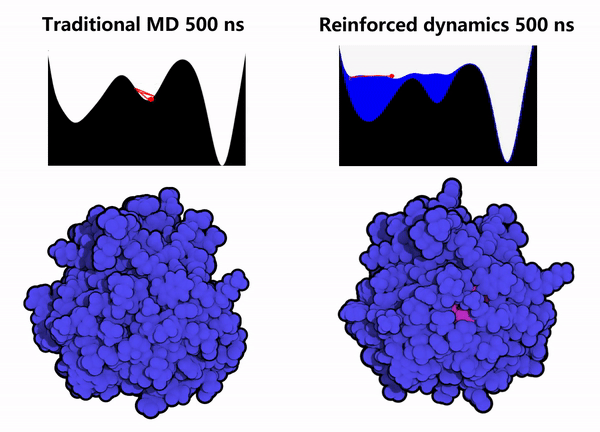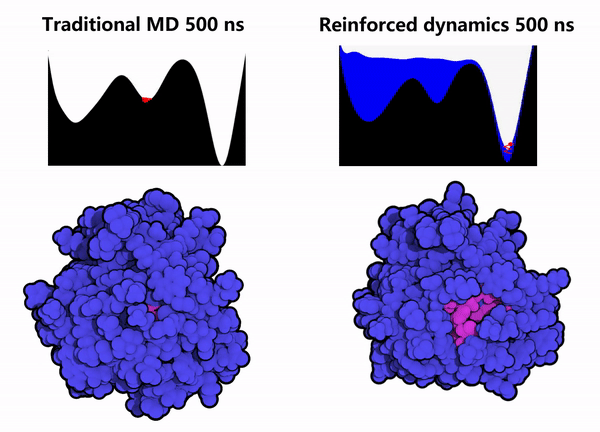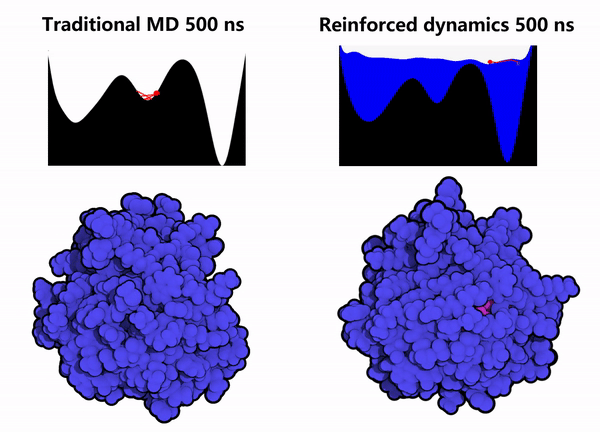
Figure 2. Enhanced dynamics enable efficient sampling of the GPX4 protein
2. Druggable Pocket Induction: In actual drug development projects, once the protein backbone structure is determined, conformational changes in side chains are also crucial. Building on the previously identified cryptic sites, the DP Technology team employed a combination of enhanced dynamics and organic solvent probes to further explore and induce this pocket. As shown in Figure 3, with the aid of organic solvent probes, we were pleased to discover that the protein surface, originally relatively flat, was induced to form a deeper small-molecule binding pocket.
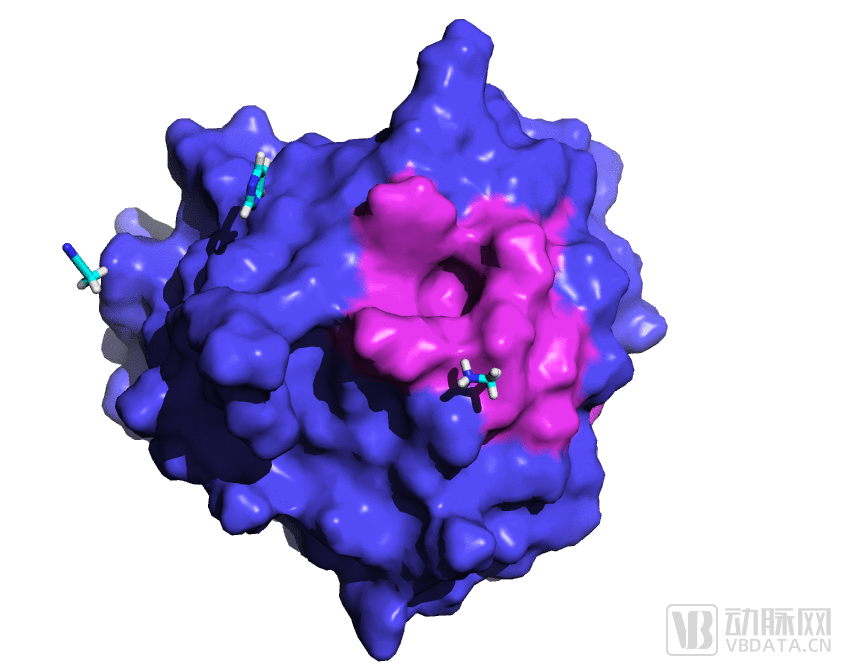
Figure 3. RiDymo-enhanced dynamics combined with the Uni-Probe organic solvent probe method induces the discovery of a novel pocket in GPX4
3. Molecular Screening and Validation: Based on this pocket, high-throughput virtual screening of molecules was performed against this site by leveraging the Uni-Docking method within DeepModeling’s Hermite drug design platform. As shown in Figure 4, the currently identified non-covalent molecules DP018 and DP029 exhibited GPX4 enzymatic inhibitory activity at the micromolar level (Figure 4A), comparable to that of the positive control covalent molecule ML162. Subsequently, surface plasmon resonance (SPR) experiments confirmed that these non-covalent molecules could directly bind to GPX4, with binding affinities also reaching the micromolar range (Figure 4B). Figure 4C illustrates the changes in intracellular reactive oxygen species (ROS) levels observed upon treatment with the DP non-covalent molecules, indicating the induction of ferroptosis.
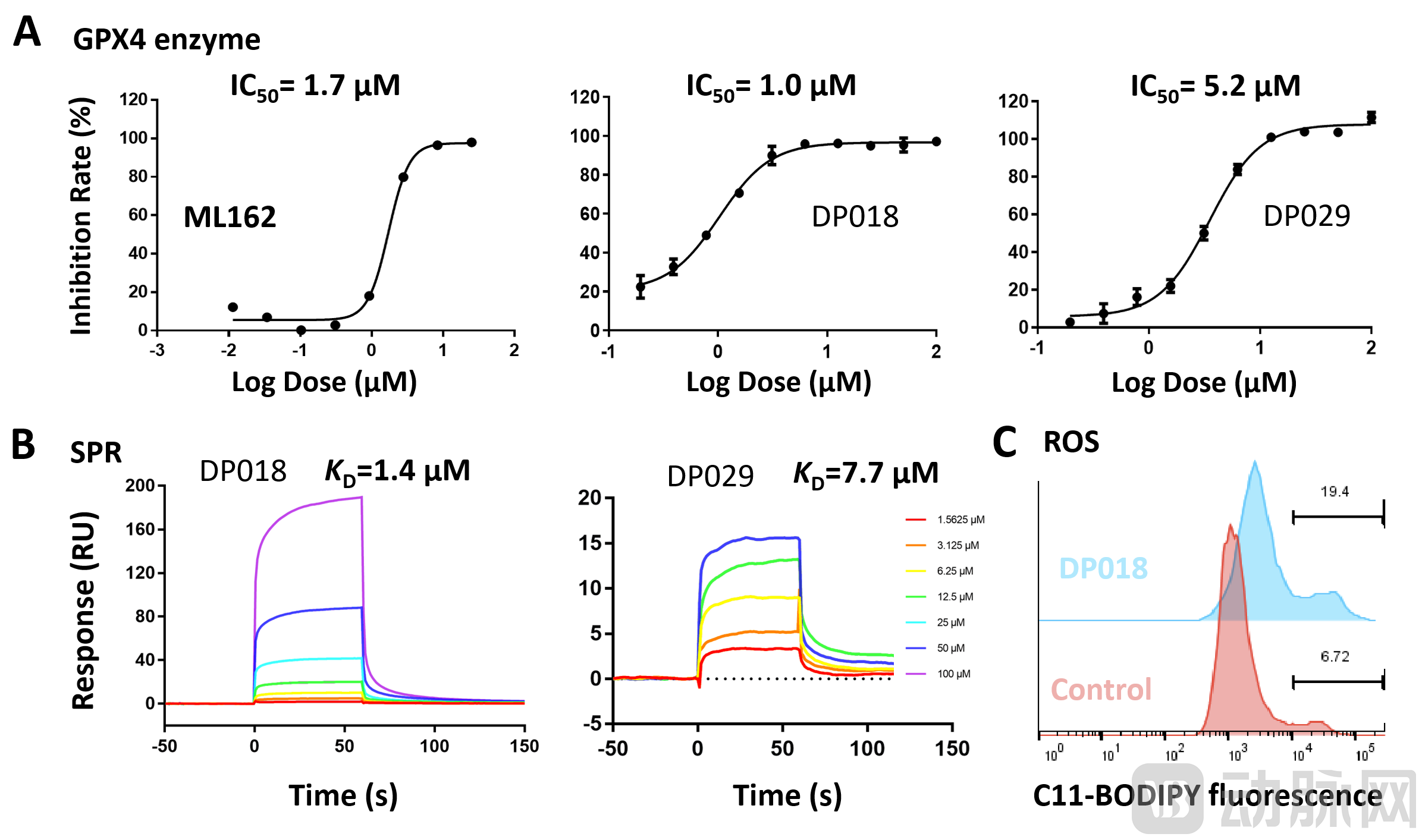 Figure 4. Results of ML162 and screened compounds in the GPX4 enzymatic assay (A), SPR assay (B), and ROS assay (C)
Figure 4. Results of ML162 and screened compounds in the GPX4 enzymatic assay (A), SPR assay (B), and ROS assay (C)
Protein dynamics research is increasingly becoming a focal point in global drug discovery and development. Conventional rational drug design typically requires target proteins to have well-defined structures and clear binding sites; such targets are commonly referred to as “druggable” targets, accounting for approximately 15% of human therapeutic targets. The remaining 85% are considered “undruggable” targets, generally characterized by flat protein surfaces lacking distinct binding pockets, high structural flexibility, or even the absence of stable three-dimensional structures. These types of targets pose significant challenges for traditional rational drug design approaches. In contrast, DeepModeling’s independently developed RiDymo enhanced dynamics platform (Figure 5) offers a novel solution to this problem.
The RiDymo platform is a dynamics-focused platform developed based on the Reinforced Dynamics (RiD) algorithm [4], specifically designed to address “undruggable” targets. Compared with other dynamics algorithms and platforms, RiDymo’s core advantage lies in its dramatically improved sampling efficiency during simulations. By fully leveraging the high-dimensional representation capabilities of neural networks, it efficiently captures dynamic conformational changes in complex biomacromolecular systems. This enables proteins to truly “move” under relatively controllable computational resources and simulation timeframes (as shown in the figure above).
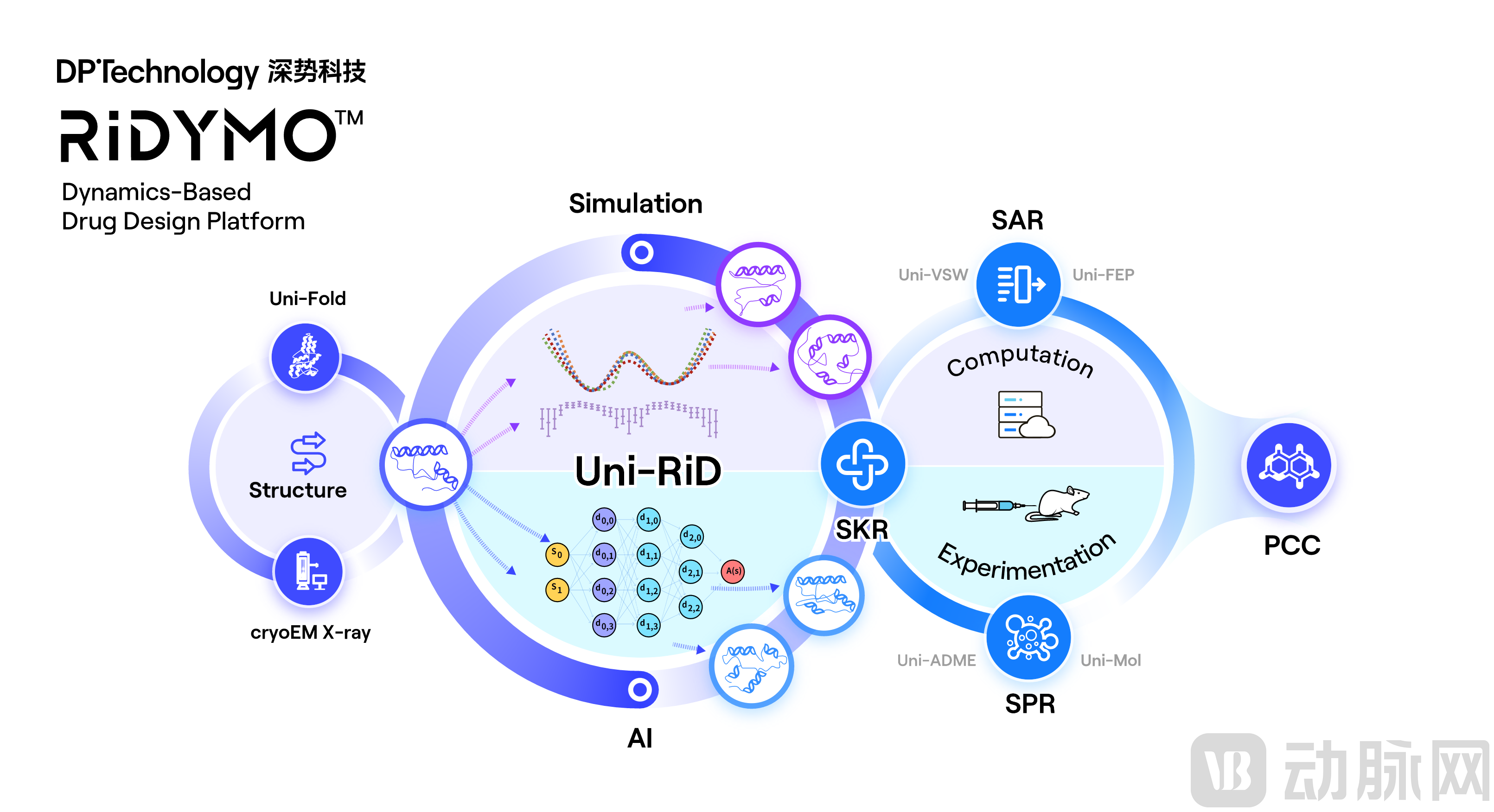 Figure 5. RiDymo Enhanced Kinetics Platform
Figure 5. RiDymo Enhanced Kinetics Platform
Previously, the RiD algorithm, a core capability of the platform, was published in a Nature subsidiary journal! DeepModeling’s RiD method addresses the challenge of high-dimensional space sampling in biomolecular systems. The article demonstrates that, for the de novo folding problem of a protein, the free energy surface obtained by the RiD method through 1.86 μs of simulation is more comprehensive than that achieved by conventional molecular dynamics methods over 100 μs, highlighting an nearly hundredfold improvement in the efficiency of molecular simulations with the RiD method.
Based on the RiD method, the RiDymo Enhanced Dynamics Platform can be used to address the following issues:
• For systems with flat surfaces or protein-protein interactions (PPIs), RiDymo can be used to induce the formation of druggable pockets and explore novel allosteric pockets in the absence of well-defined binding sites.
• For systems with significant structural dynamics, such as GPCRs and ion channels, RiDymo can analyze residue correlations along motion trajectories, facilitating the study of drug-target mechanisms and drug screening.
• For intrinsically disordered proteins that lack stable structures in vivo, RiDymo can explore their important conformations in terms of thermodynamics and kinetics, and identify druggable structural features.
• Furthermore, since drugs ultimately need to exert their effects in vivo, considering only the thermodynamic parameter of binding affinity is insufficient. RiDymo can effectively calculate kinetic parameters that are strongly correlated with in vivo drug efficacy, such as residence time (i.e., the reciprocal of the dissociation rate constant), an aspect that has often been overlooked in the past.
The RiDymo platform is dedicated to establishing structure-kinetic relationships (SKR) in drug molecule discovery, efficiently advancing the identification of high-quality preclinical candidate compounds by closely integrating structure-activity relationships (SAR) and structure-property relationships (SPR).
The disclosure of pipeline progress in this instance fully validates the immense potential of DeepModeling’s RiDymo platform in real-world drug development, serving as a comprehensive demonstration of the “AI for Science” new paradigm transitioning from algorithms to tools and ultimately to practical applications.
We believe that as research into the mechanisms of ferroptosis becomes more comprehensive in the future, drug development strategies targeting related pathways will also become more robust. Leveraging DeepModeling’s profound understanding of molecular dynamics, we will continue to overcome challenges associated with difficult-to-drug targets and achieve more remarkable results across a broader pipeline.
About the Drug R&D Team at DP Technology
DP Technology’s drug discovery team, guided by the new paradigm of “AI for Science,” focuses on leveraging advanced computational methods that integrate artificial intelligence with molecular simulations to drive drug development against difficult-to-drug targets. Led by Dr. Zhang Xiaomin, the core team members possess strong interdisciplinary backgrounds and extensive experience in drug development. By leveraging DP Technology’s globally leading enhanced sampling platform, RiDymo, and its de novo design platform for macromolecules, the team has established a comprehensive R&D system through a combination of self-developed pipelines and collaborative development, covering three major therapeutic areas: central nervous system (CNS) disorders, oncology, and autoimmune diseases.
BD Collaboration: bd@dp.tech
PR Inquiries: pr@dp.tech
Reference
[1] Stockwell, B. R. (2022). Ferroptosis turns 10: Emerging mechanisms, physiological functions, and therapeutic applications. Cell, 185(14), 2401-2421.
[2] Wang, F., & Min, J. (2021). DHODH tangoing with GPX4 on the ferroptotic stage. Signal Transduction and Targeted Therapy, 6(1), 1-2.
[3] Moosmayer, D., Hilpmann, A., Hoffmann, J., Schnirch, L., Zimmermann, K., Badock, V., ... & Hillig, R. C. (2021). Crystal structures of the selenoprotein glutathione peroxidase 4 in its apo form and in complex with the covalently bound inhibitor ML162. Acta Crystallographica Section D: Structural Biology, 77(2), 237-248.
[4] Wang, D., Wang, Y., Chang, J., Zhang, L., Wang, H. & E, W. (2022). Efficient sampling of high-dimensional free energy landscapes using adaptive reinforced dynamics. Nature Computational Science, 2(1), 20-29.