NMPA Classifies Certain Digital Therapeutics as Class III Medical Devices in Latest Classification Guidance

SLANHEALTH
Digital Therapeutics CDMO and Full-process Service Developer
On March 30, the Center for Medical Device Standardization Administration of the National Medical Products Administration (hereinafter referred to as the Standardization Center) released the Summary of the First Batch of Medical Device Classification and Determination Results in 2023. Among them, two types of software medical devices that meet the definition of digital therapeutics—“Software for Assessment and Treatment of Cognitive Impairment” and “Insulin Calculation Software”—were classified as Class III medical devices.
Does this mean that regulation of these two categories of digital therapeutics will become stricter? VCBeat (WeChat ID: VCBeat) interviewed industry experts to explore the underlying reasons.
As is well known, medical devices are subject to classified management based on their safety and efficacy. The International Medical Device Regulators Forum (IMDRF) is a voluntary regulatory harmonization organization led by medical device regulatory authorities from countries around the world, including the National Medical Products Administration (hereinafter referred to as the NMPA).
The IMDRF classifies medical devices into four categories based on risk: Class A (low risk), Class B (low-to-moderate risk), Class C (moderate-to-high risk), and Class D (high risk). In addition to directly adopting the IMDRF’s four-tier classification system, some countries and regions have made corresponding adjustments based on their national circumstances and specific objectives.
The classification and management of medical devices are primarily based on product risk.
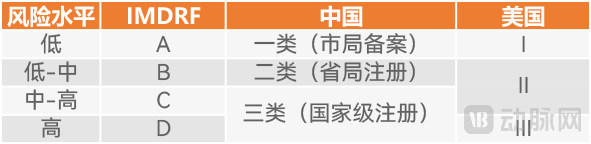
For instance, both China and the United States classify medical devices into three categories. The difference lies in the fact that medium- to high-risk medical devices are classified as Class II in the United States, whereas they are classified as Class III in China. This clearly indicates that, solely in terms of medical device classification, China’s regulatory standards are more stringent. Consequently, due to this stricter regulation, the costs associated with market entry and subsequent commercialization for these medical devices are higher.
In 2017, the NMPA issued the Catalogue of Medical Device Classification to replace the Rules for Medical Device Classification issued in 2000, thereby serving as the guidance for formulating China’s medical device classification catalogue and determining the regulatory categories for new medical devices. The Catalogue classifies medical devices into three classes.
Class I (low-risk) medical devices are those for which routine administration is sufficient to ensure their safety and effectiveness; these are currently approved by municipal drug regulatory authorities. Class II (medium-risk) medical devices are those whose safety and effectiveness must be controlled; these are currently approved by provincial drug regulatory authorities. Class III (high-risk) medical devices are those implanted into the human body to support or sustain life, pose potential risks to the human body, and require strict control over their safety and effectiveness; these are currently approved by the National Medical Products Administration.
In essence, the "Classification Catalog" covers nearly all medical devices available on the market at that time. However, it is evident that no catalog can encompass the continuous influx of newly emerging devices. Therefore, the NMPA also issues announcements from time to time to adjust the "Classification Catalog."
For example, in the March 2022 adjustment to the “Classification Catalog,” RF therapy devices (non-ablation) and RF skin therapy devices, which had previously been classified as Class II medical devices, were reclassified as Class III medical devices. Relevant enterprises were required to obtain new medical device registration certificates in accordance with the updated regulations by April 1, 2024; otherwise, they would be prohibited from manufacturing, importing, and selling these products.
The original intent of this adjustment was to standardize and regulate the “Thermage” aesthetic procedure, which was plagued by widespread irregularities at the time. Meanwhile, the new regulations further raised market entry barriers, accelerating consolidation within the energy-based medical aesthetics industry. This illustrates the significant impact of reclassifying medical devices.
However, even with irregular adjustments, the system still fails to meet the needs of the rapidly growing medical device industry. New products under research and development may not be found in the "Classification Catalog," or their definitions and classifications may be ambiguous, making it difficult to determine their proper classification. This poses significant obstacles to product commercialization and development, and may even threaten the very survival of enterprises.
For such cases, the NMPA has also established a specialized classification determination procedure. If it is difficult to determine the classification of a product, the relevant working group under the NMPA headquarters will conduct a classification determination to clarify the regulatory category applicable to the product.
VCBeat’s statistics reveal that since 2018, the Standardization Management Center has annually released a summary of the classification and determination results for medical devices. In recent years, the frequency of these releases has increased, with three such summaries published in 2022 alone.
In the summary of results released on March 30, two software medical devices that fit the definition of digital therapeutics—namely, the “Cognitive Impairment Assessment and Treatment Software” and the “Insulin Calculation Software”—were prominently featured. These represent two typical categories of digital therapeutics for mental health and diabetes management, respectively, and both have been classified as Class III medical devices. This development has sparked widespread discussion within the industry, with many suggesting that it may signal a future regulatory trend wherein these two types of software will be managed as Class III medical devices.
Since digital therapeutics in China have historically been approved primarily as Class II medical devices, reclassification as Class III devices would entail significantly stricter regulatory oversight. This poses considerable challenges for most digital therapeutics companies in their early startup stages.
As a specialized CDMO and full-process service platform for digital therapeutics (DTx), SLANHEALTH has established an end-to-end service system encompassing DTx project assessment, integrated design and R&D development, pilot-scale translation, registration-related clinical trials, product registration, post-market support, and real-world studies. Anny, the head of regulatory affairs at SLANHEALTH, is highly knowledgeable about registration matters. She stated in an interview with VCBeat: “The classification determination process follows a ‘case-by-case’ approach. For each classification review, the Classification Review Committee, composed of experts from various fields, discusses and ultimately determines the classification opinion based on the specific characteristics of the product. The classification summary issued by the Standardization Management Center merely aggregates classification opinions provided by provincial and national authorities within the classification determination system over a certain period; it is not a regulatory document. Therefore, while it offers some reference value for the classification of similar products in the future, it is neither absolute nor legally binding. Furthermore, the product descriptions included in the classification summary are too brief to adequately determine whether products belong to the same category.”
“Only the latest version of the Classification Catalogue issued by the National Medical Products Administration, along with related classification determination guidelines, serves as the ‘gold standard’ used by drug regulatory authorities for device classification and determination,” she added.
VCBeat has compiled a summary of classification and determination results from 2018 to the present. Taking “cognitive function” as a keyword example, in addition to the current “Software for Assessment and Treatment of Cognitive Impairment,” a “Software for Cognitive Function Assessment and Training” was classified as a Class III medical device in 2022. Earlier, in 2020, a “Software for Treatment of Cognitive Impairment” and an “Auxiliary Assessment System for Brain Physiology and Cognitive Function” were classified as Class II medical devices.
Also in 2022, two medical devices obtained Class II medical device registration certificates under the name “Cognitive Function Assessment and Training Software.” This illustrates the case-by-case nature of classification determinations.
For digital therapeutics, classification as Class III devices is not as severe as one might imagine.
“The anticipated risk of a product is the key factor in determining whether a medical device is classified as Class II or Class III. For instance, this includes assessing whether the disease type, disease progression, and target population pose higher risks, such as critically ill patients or those experiencing acute episodes. Another consideration is the role of software-generated output in clinical practice. Furthermore, the inclusion of algorithms, such as those for artificial intelligence or drug dosage calculations, is a characteristic feature of Class III medical devices,” Anny explained to us regarding the principles of medical device classification.
Based on the summarized results, the product description for “Insulin Calculation Software” is as follows: It reads blood glucose data from glucometers with data communication capabilities for adult patients with poorly controlled diabetes. By combining this data with entered personal information (including age, height, weight, C-peptide levels, etc.), and leveraging diabetes guidelines/expert consensus along with artificial intelligence algorithm models, it determines the timing of insulin administration and the corresponding insulin formulation. The software is used to recommend insulin formulations and dosage adjustments; physicians may refer to the recommended insulin dosage adjustments when formulating treatment plans.
Among these, the features “Artificial Intelligence” and “Insulin Dosage Adjustment Regimens for Physician Reference” vividly demonstrate the product’s use of AI for clinical decision support. The Guiding Principles for the Classification of Medical Artificial Intelligence Software clearly stipulate that medical software used to provide diagnostic conclusions or perform drug dosage calculations shall be regulated as Class III medical devices.
Its classification code is categorized under 21-04, which corresponds to the category of decision support software. In the "Classification Catalog," drug calculation software and decision support software that provide clinical diagnostic and therapeutic basis and/or recommendations are also explicitly stipulated to be regulated as Class III medical devices.
In the description of the “Cognitive Impairment Assessment and Treatment Software,” it is also mentioned that assessment results are derived by analyzing cranial magnetic resonance imaging (MRI) data, and dynamic adaptive training and treatment plans are provided. According to the “Classification Catalog,” decision-support software in which computer programs automatically identify lesions and provide clinical diagnostic and therapeutic bases and/or recommendations regarding the nature of such lesions is also regulated as a Class III medical device.
Furthermore, this issue’s summary includes a “Clinical Information System for Anesthesia and Surgery” classified under Class III regulation. While the name alone may be difficult to interpret, the product description features phrases such as “storing, recording, displaying, processing, and providing early warnings for data, and offering risk assessment algorithmic models to assist physicians in evaluating patients’ anesthesia risks,” as well as “assisting anesthesia and surgical healthcare professionals in patient risk assessment.” These descriptions align with the characteristics of “AI + Clinical Decision Support.”
Among the historical summaries of classification determinations, only five Class II medical devices were categorized as Class II management for 21-04 decision support software.
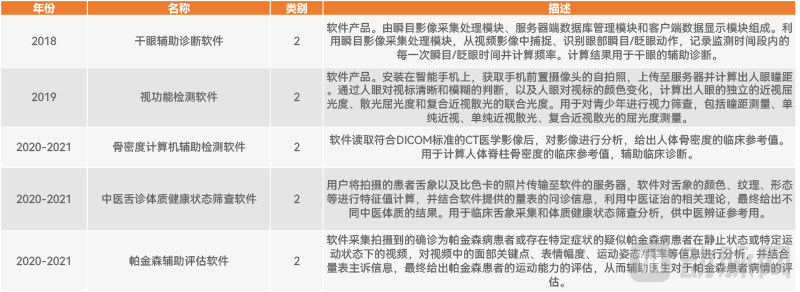
VCBeat’s analysis of historical classification and boundary determination results reveals that once a product description includes “artificial intelligence + decision support,” it is generally classified under 21-04 Decision Support Software. Among the 61 products categorized as 21-04 Decision Support Software, only five with significantly lower risk levels are regulated as Class II medical devices. These are: Dry Eye Auxiliary Diagnosis Software (2018), Visual Function Testing Software (2019), Computer-Aided Bone Mineral Density Detection Software (2020–2021), Traditional Chinese Medicine Tongue Diagnosis and Constitutional Health Status Screening Software (2020–2021), and Parkinson’s Disease Auxiliary Assessment Software (2022).
In addition, Anny stated that the classification of the “Cognitive Impairment Assessment and Treatment Software” as a Class III medical device is also attributable to China’s relatively stringent regulatory oversight of psychiatric and psychological medical devices.
As previously mentioned, classification determinations are conducted on a case-by-case basis and hold mandatory force only for the specific products under evaluation. Therefore, while the designation of several products as Class III medical devices in this instance provides reference value for the classification of similar future products, it cannot serve directly as the definitive basis for classifying such products.
“As the number of medical devices of the same type requiring classification determination gradually increases, regulatory authorities will also consider making formal amendments to the Classification Catalog or issuing corresponding guidelines for classification determination. Only when the regulatory authorities officially issue documents to clearly change the management category of a certain class of medical devices do affected approved products need to undergo registration changes to meet new regulatory requirements. The transition period in between is generally sufficient to accommodate the time required for enterprises to complete registration changes,” Anny explained to VCBeat.
“If no official document is issued, such a summary of classification determinations will not have any impact on other approved Class II medical devices in the same therapeutic area from a compliance perspective. Of course, from a marketing standpoint, if a product ultimately obtains approval under Class III regulatory management, it will certainly exert some influence on competing Class II products, for instance, by emphasizing the authority of Class III certification, as well as the clinical efficacy and safety of the product during promotion.”
VCBeat has found that most products classified as Class III medical devices through the classification determination process ultimately fail to complete registration and reach the market. This is not surprising, as medical devices entering the classification determination process are somewhat different from existing products and possess a certain degree of innovativeness. However, it remains uncertain whether such innovations can meet the regulatory requirements for efficacy and safety of medical devices.
Furthermore, the barriers to obtaining a Class III medical device registration certificate are significantly higher than those for a Class II certificate, in terms of both capital requirements and time investment. This may prove unrealistic for startups that originally planned to secure a Class II certification. In such cases, it is more likely that companies will opt to improve existing products by eliminating certain high-risk features, thereby facilitating faster regulatory approval.
In highly regulated sectors such as medical devices, any uncertainty can delay time-to-market. Therefore, the establishment of relatively comprehensive classification and definition guidelines would significantly alleviate this issue. Taking medical AI as an example, the release of the Guiding Principles for Product Classification and Definition of Artificial Intelligence Medical Software has brought greater clarity and operational feasibility to the definition, categorization, and regulation of AI-based medical software products, providing clear guidance for companies in the industry regarding product positioning and registration strategies.
Subsequently, the “Guiding Principles for Registration Review of AI Medical Devices” was released, providing a detailed description of concepts, basic registration principles, lifecycle processes, technical considerations, and other aspects of AI medical devices, thereby offering clear regulations and standards for registering enterprises.
This facilitated the rapid development of AI-based medical devices in 2022, with a total of 25 Class III AI medical device approvals granted—the highest annual number on record.
For the digital therapeutics industry, the good news is that corresponding regulatory detailed rules are currently being formulated. Guidelines for the classification and definition of digital therapeutics as medical devices are under development; once completed, they will clarify the definition, categorization, and regulation of digital therapeutics medical devices, thereby providing greater clarity for digital therapeutics companies in terms of product positioning and registration.