Zhu Baolin of Puhua Capital: Opportunities in Next-Generation Conjugate Therapeutics Beyond ADCs
Editor's Note: This article is sourced fromPuHua Capital, AuthorZhu Baolin, reprinted with permission from VCBeat
In addition to the more mature ADCs, FDCs, and RDCs, areas poised for breakthrough advancements, driven by improvements in conjugation technologies and a deeper understanding of molecular types, include protein degradation conjugates and antibody-nucleotide conjugates. Of course, beyond scientific considerations, market conditions and competitive advantages must also be taken into account, butThere is still significant potential for the emergence of next-generation “ADCs.”
In March 2023, Pfizer acquired Seagen for $43 billion. In early April, Duality Biologics became another Chinese ADC innovator to secure an upfront payment exceeding $100 million, following RemeGen and Kelun Pharmaceutical. Looking back at the past few years, M&A activity in the global ADC market has remained robust. What accounts for this strong appeal to industry giants? Beyond the iterative optimization of individual ADC components, where lies the next “ADC”?
Number and Types of ADC Transactions:
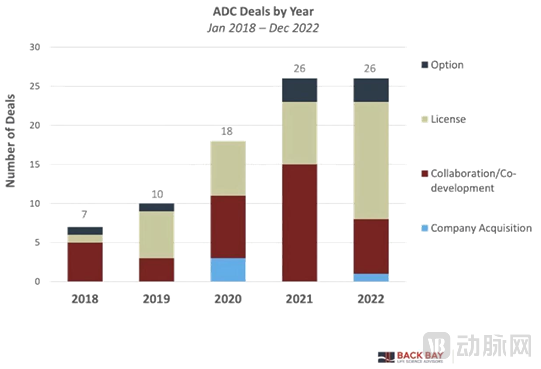
Since 2018, the transaction volume of ADC-related assets has more than tripled, with licensing and co-development models accounting for the largest share.
Number of ADC Transactions by Clinical Stage:
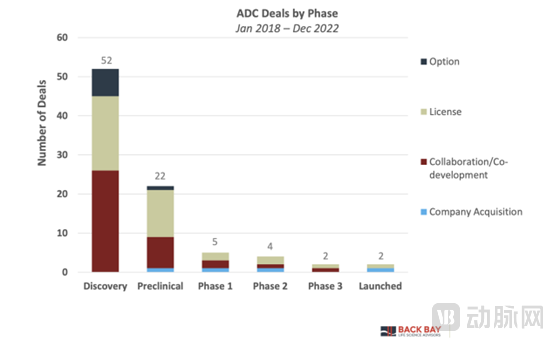
ADC deals are frequently struck early in the product development lifecycle. This underscores the significant interest in early-stage technologies and assets, particularly during the discovery phase, which involves monoclonal antibody selection, linker optimization, and toxin payload selection.
Comparison of Average Upfront and Milestone Payments for ADCs versus Other Molecular Modalities:
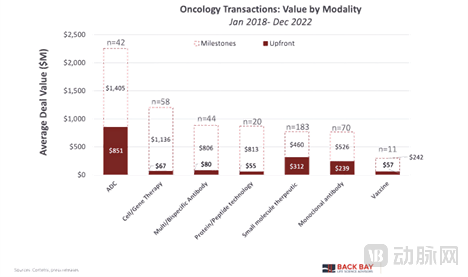
Although antibody-drug conjugates (ADCs) are involved in fewer transactions than certain other molecular formats, they command the highest transaction values. On average, upfront payments across all development stages amounted to $851 million (based on 42 disclosed deals), with five of these transactions featuring upfront payments of $1 billion. In terms of median total deal value, ADCs demonstrated an overall value of $848 million, which is $300 million higher than the median value of $512 million observed for multi-/bispecific antibodies.
Transaction Volumes for ADCs Across Different Clinical Stages:
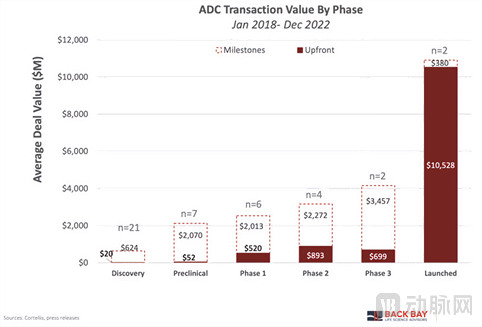
The most striking aspect of the ADC deal landscape is the total transaction value across various development stages. Although the total transaction value has continued to rise from preclinical through Phase 3 clinical trials (from approximately $2.5 billion in the preclinical stage to around $4 billion in Phase 3), upfront payments have increased significantly (from $52 million in the preclinical stage to $520 million in Phase I). This trend underscores the value derived from de-risking druggability through clinical data.
Antibody-drug conjugates (ADCs) have a development history spanning nearly a century, with the first genuine attempt dating back to 1958, when Mathe conjugated anti-mouse leukocyte immunoglobulin with methotrexate for the treatment of leukemia. However, this initial effort ended in failure. It was not until 2000 that the first ADC drug, gemtuzumab ozogamicin, was officially launched. The second ADC, brentuximab vedotin, entered the market in 2011, followed by the approval of more than ten additional agents over the subsequent 11 years. Overall, ADCs only truly entered a phase of rapid development starting in 2011. Unlike many cutting-edge therapies that receive critical acclaim but struggle with commercial adoption, ADCs have been unlocking substantial commercial value over the past five years, driven by their transformative clinical efficacy and broad range of indications. Since its launch in late 2019 as a last-line therapy, trastuzumab deruxtecan (DS-8201) has continuously expanded into earlier lines of treatment and HER2-low expression populations, demonstrating remarkable commercial momentum. Its global sales reached $1.26 billion in 2022, and Nature projects that its sales will reach $6.2 billion by 2026.
(1) Structure Determines Properties
Antibody-Drug Conjugates (ADCs) consist of three components: a monoclonal antibody that targets cancer cells, a highly bioactive small-molecule drug, and a linker that connects the monoclonal antibody to the small-molecule drug.
Following binding to cell-surface antigens on cancer cells, antibodies mediate the internalization of antibody–drug conjugates (ADCs) via endocytosis. The ADCs are then transported through endosomes to lysosomes, where linker or antibody degradation occurs, resulting in the release of cytotoxic small-molecule drugs that kill tumor cells.
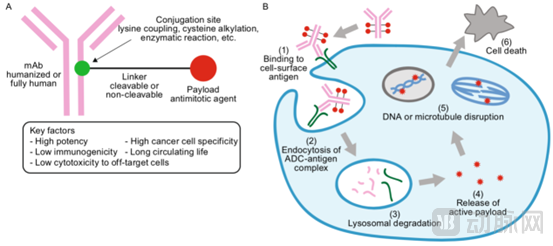
The core of ADC drug development lies in four key elements: antibodies, linkers, small-molecule toxins, and conjugation technologies.
1.1 Antibody-related aspects involve target selection and antibody type selection
Target Selection:
Ideal targets can yield lower non-specific toxic side effects and a better therapeutic index. Typically, selectable target antigens include tumor-specific antigens (TSAs) expressed in tumor tissues, and tumor-associated antigens (TAAs) that are highly expressed in tumor tissues but lowly expressed in normal tissues. In addition to specific expression requirements, these targets must also mediate endocytosis to enable intracellular activity of small-molecule drugs. Studies have shown that ADC activity correlates with the number of antigens on the cell surface; at least 10^4 surface antigens are required to ensure delivery of a lethal dose of cytotoxic drug into the cell for ADC efficacy. However, the clinical success of DS-8201 in HER2-low indications has broadened perspectives for future target selection.
ADCs Offer a Wide Range of Target Options:
− Antibody targets with approved drugs (e.g., Her2);
− High-expression, internalizable, and mechanistically well-understood undrugged targets at the target site (e.g., folate receptor-alpha/FR-α);
− Undruggable targets whose mechanisms have not been fully validated
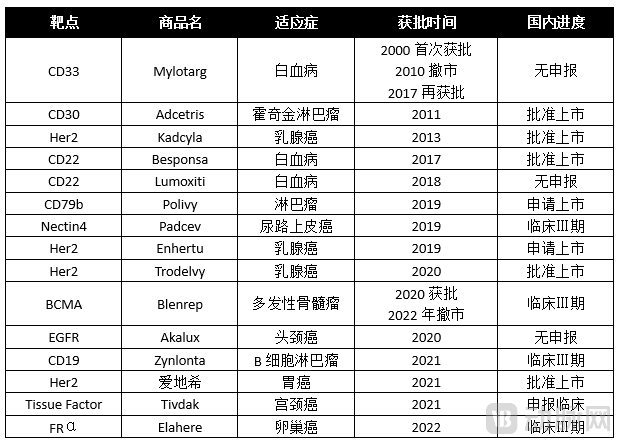
Fifteen marketed ADC drugs target 11 distinct antigens: CD33, CD30, HER2, CD22, CD79b, Nectin-4, BCMA, EGFR, CD19, Tissue Factor, and FRα.
According to statistics from the Yaozhi Network and PharmaCube database, among the 425 ADC drugs currently under development, the more prominent targets include HER2, EGFR, CLDN18.2, TROP2, c-Met, CD19, PSMA, Muc1, BCMA, and PD-L1, most of which are well-validated and mature targets.
Further ADC targets awaiting validation include:
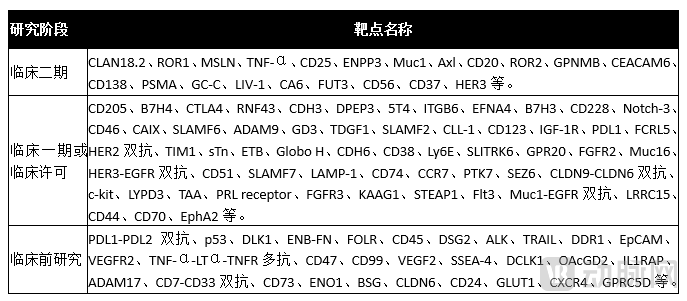
Note: Compiled from public information of Sino Biological.
Antibody Selection:
The selection of the antibody variable region is highly correlated with the target, requiring high specificity and strong target-binding affinity. The selection of the constant region requires low immunogenicity and low cross-reactivity to achieve more efficient uptake of ADC drugs by tumor cells and a longer half-life of ADC drugs in serum.
− The selection of antibody sources for ADCs has undergone roughly four stages: murine, chimeric, humanized, and fully human antibodies. Due to issues such as high immunogenicity, low efficacy, and short circulation half-life associated with murine and chimeric antibodies to varying degrees, humanized and fully human antibodies have become the preferred choice in ADC drug design with the advancement of antibody and genetic engineering technologies. Currently, the vast majority of clinical and preclinical studies utilize these latter two types of antibodies.
− In addition to antibody origin, human antibodies comprise multiple subtypes. Among these, IgG1 is the most abundant in serum and offers an optimal balance between a long half-life and potent immune activation, making it the most extensively studied and utilized antibody format for antibody-drug conjugates (ADCs). Besides IgG1, IgG4 is also frequently employed in the design of ADC drugs that require minimal immunogenic responses, owing to its reduced immune activation effects. Both IgG1 and IgG4 possess 12 intra-chain disulfide bonds and 4 inter-chain disulfide bonds; the inter-chain disulfide bonds, characterized by high reactivity, are commonly used as conjugation sites for linkers. However, due to the propensity of IgG4 for Fab-arm exchange, which can lead to off-target effects, the S228P mutation strategy is often implemented in ADC development using IgG4 antibodies to mitigate Fab-arm exchange.
− Furthermore, the inherent signaling functions of the antibody itself, such as antibody-dependent cellular cytotoxicity (ADCC), antibody-dependent cellular phagocytosis (ADCP), and complement-dependent cytotoxicity (CDC), should be carefully considered. ADCC and ADCP mediate immune system involvement in the killing of tumor cells, thereby influencing the ultimate therapeutic efficacy of ADCs. Therefore, thorough investigation of the antibody’s ADCC and ADCP effects is essential during the design of ADC drugs.
1.2 Selection of Small-Molecule Toxins (Payloads):
Toxin molecules are a key factor in the successful development of antibody-drug conjugate (ADC) therapeutics, directly determining the cytotoxic effect on target cells.
Early research on antibody-drug conjugates (ADCs) generally selected traditional small-molecule chemotherapeutic agents, including doxorubicin. However, only a small fraction of the injected antibodies accumulated in solid tumor tissues, and the number of small-molecule drugs conjugated to each antibody was limited. Consequently, small-molecule drugs with half-maximal inhibitory concentration (IC50) values in the micromolar range failed to achieve satisfactory therapeutic efficacy. Currently, the selection of small-molecule payloads often requires IC50 values as low as the nanomolar or even picomolar range. These small-molecule drugs primarily fall into two categories: tubulin inhibitors and DNA-damaging agents. Furthermore, the cytotoxic payload must possess suitable functional groups for conjugation, exhibit potent cytotoxicity, demonstrate hydrophobicity, and maintain high stability under physiological conditions.
In addition to requiring a low IC50 value, small-molecule drugs are generally expected to possess the following attributes: 1) they should not readily induce aggregation of the antibody-drug conjugate (ADC) after conjugation with the antibody, thereby ensuring a prolonged circulation time in vivo; 2) they should exhibit low immunogenicity both as standalone entities and when incorporated into ADCs; 3) they should demonstrate sufficient stability in aqueous solutions and blood, while possessing suitable reactive sites for conjugation; 4) they should retain their biological activity after being conjugated to the antibody via a linker; and 5) they should be synthesizable through relatively cost-effective processes.
1.3 Selection of Linkers:
Although selecting specific antibodies and payloads based on the type of tumor cells is important, linkers can significantly influence the toxicity, stability, specificity, and other properties of the final antibody-drug conjugate (ADC) in terms of pharmacokinetics, pharmacodynamics, and therapeutic window. Therefore, constraining the antibody and payloads by choosing an appropriate linker is key to successfully constructing an ADC. An ideal linker must meet the following conditions: 1) The linker needs to remain stable in the bloodstream but rapidly release active payloads when localized within or near tumor cells; instability of the linker may lead to premature payload release, causing damage to normal tissue cells; 2) Once the ADC is internalized into target tumor tissues, the linker should be capable of rapid cleavage and release of toxic molecules; 3) Hydrophilicity/hydrophobicity is also an important characteristic to consider for linkers, as hydrophobic linker groups and hydrophobic payloads often cause aggregation of ADC small molecules, thereby inducing immunogenicity.
Linkers can be broadly categorized into non-cleavable linkers, predominantly used in first-generation antibody-drug conjugate (ADC) therapeutics, and cleavable linkers, which are more commonly employed in second- and third-generation ADC drugs.
Non-cleavable Type:
Non-cleavable linkers primarily include maleimide-based and thiol-containing hexanamide-based linkers, which conjugate small-molecule drugs to antibodies by forming amide bonds and thioether bonds.
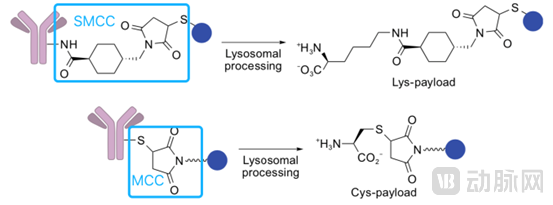
Non-cleavable linkers ensure high stability of antibody-drug conjugates (ADCs) during systemic circulation. After entering cells via antibody-antigen-mediated internalization and endosomal transport to lysosomes, the antibody is cleaved by various lysosomal enzymes, releasing the small-molecule drug and exerting cytotoxic effects. However, ADCs employing non-cleavable linkers typically target tumor cells with high antigen expression while having minimal impact on surrounding tumor cells with low antigen expression. This is because the small-molecule drugs released after enzymatic degradation in lysosomes usually retain charged amino acid residues. These charged amino acid–small molecule complexes cannot cross the cell membrane, thereby preventing the killing of adjacent tumor cells through the bystander effect.
Cleavable Type:
Cleavable linkers are primarily categorized into pH-sensitive (hydrazone, carbonate), enzyme-sensitive (peptide, β-glucuronide), and reducible (disulfide bond) types, leveraging the environmental differences between the systemic circulation and tumor cells. These linkers undergo cleavage in response to the lower pH within acidic endosomes (pH 5.0–6.0) or lysosomes (pH 4.8) of tumor tissues, intracellular proteases, or the higher reducing potential inside tumor cells. Antibody-drug conjugates (ADCs) employing such linkers achieve targeted delivery to tumor cells via antibody-mediated localization, accumulate in the tumor region, and release small-molecule drugs upon stimulation by the unique tumor microenvironment, thereby inhibiting cell proliferation and inducing cytotoxicity. Compared with non-cleavable linkers, cleavable linkers exhibit lower dependence on internalization by tumor cells, which facilitates the bystander killing effect of ADCs.
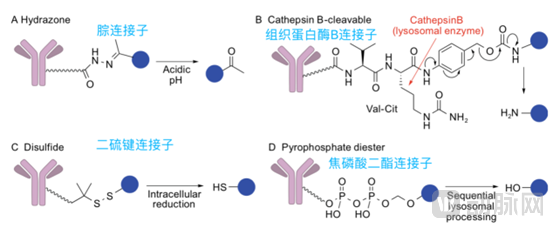
It is evident that the choice of linker type is closely related to target selection. In ADC drugs with cleavable linkers, targets identified as B-cell antigens (CD19, CD20, CD21, CD22, CD79B, CD180) have been proven highly effective in vivo. Conversely, in ADC drugs with non-cleavable linkers, targets such as CD22 and CD79b, which are confirmed to undergo endocytosis and rapid transport to lysosomes in vivo, are effective.
1.4 Conjugation Technology
Conjugation methods can be classified into chemical conjugation and enzymatic conjugation, as well as site-specific conjugation and non-site-specific conjugation.
Chemical conjugation includes: lysine amide conjugation, cysteine conjugation, and conjugation via incorporation of non-natural amino acids.
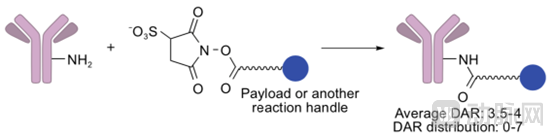
Lysine Amide Coupling:
Amino groups on lysine residues can be linked to linkers containing active carboxylic esters via amide bonds, thereby enabling further conjugation with drugs. Due to the abundance of lysine residues on the antibody surface, the resulting antibody-drug conjugates (ADCs) will comprise a heterogeneous mixture of conjugates with varying drug-to-antibody ratios (DARs) and diverse conjugation sites. After optimization, the optimal mean DAR is 3.5–4, with an actual distribution ranging from 0 to 7, indicating poor homogeneity. Furthermore, since certain lysine residues play critical roles in antigen-antibody interactions, this method may reduce antibody affinity.
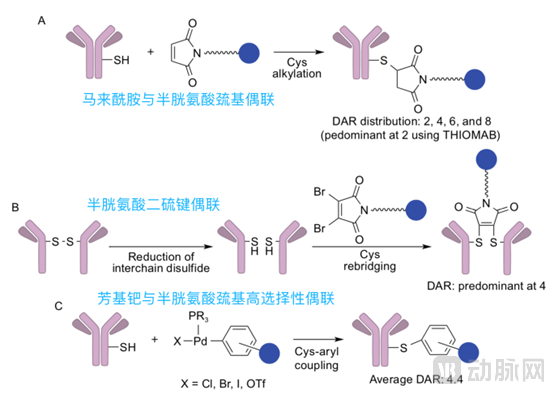
Cysteine coupling includes:
1) Traditional Cysteine Conjugation: Under normal circumstances, cysteine residues in antibodies contain sulfhydryl groups both interchain and intrachain, which exist in the form of disulfide bonds. Interchain disulfide bonds have minimal impact on antibody properties and can be cleaved using mild reduction methods to generate free sulfhydryl groups. In IgG1, there are four interchain disulfide bonds and twelve intrachain disulfide bonds. The interchain disulfide bonds can be selectively reduced to yield 2, 4, 6, or 8 free sulfhydryl groups for subsequent conjugation. Due to the limited number of conjugation sites, this method is superior to lysine amide conjugation; however, its homogeneity still requires improvement.
2) Engineered cysteine conjugation (ThioMab technology): Site-directed mutagenesis is used to artificially introduce cysteine residues into monoclonal antibodies for conjugation. The resulting ADCs have a mean drug-to-antibody ratio (DAR) of 1.9, antibody homogeneity greater than 90%, and demonstrate activity both in vitro and in vivo;
3) Cysteine disulfide bond rebridging conjugation: Reagents such as dibromomaleimide can be used to form rebridges between reduced interchain sulfhydryl groups, achieving site-specific conjugation, with the resulting drug-to-antibody ratio (DAR) primarily being 4;
4) Highly Selective Arylpalladium Coupling: By employing arylpalladium for highly selective coupling, the resulting drug-to-antibody ratio (DAR) is predominantly 4.4. This method does not require a linker, and the product exhibits stability against acids, bases, and oxidation; however, the use of palladium catalysts necessitates consideration of issues such as toxicity, cost, and palladium residue.
The first method is non-site-specific conjugation, while the latter three methods are site-specific conjugation.
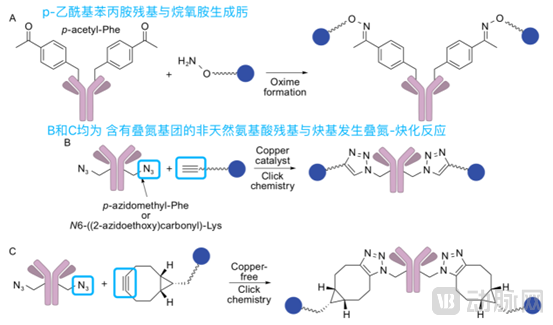
Incorporation of Non-Natural Amino Acid Conjugation:
Introduction of non-natural amino acids and their utilization for conjugation via characteristic functional groups, including methods such as:
1) A specialized codon-tRNA synthetase can be used to incorporate p-acetylphenylalanine residues containing a carbonyl group into proteins; the carbonyl group then reacts with an alkoxyamine to form an oxime, enabling further conjugation with drugs.
2) Introduce non-natural amino acid residues containing azide groups, such as p-azidomethylphenylalanine, which can be linked to alkynyl groups via azide-alkyne cycloaddition. By modifying the structure of the alkyne, this reaction can proceed both in the presence and absence of copper catalysis.
Insertion of non-natural amino acid conjugation belongs to site-specific conjugation technology. The resulting ADCs exhibit good homogeneity, but this method requires specialized techniques, and the immunogenicity of these non-natural amino acids is unknown, so safety considerations are necessary.
Enzyme coupling includes:
Transpeptidase-catalyzed transpeptidation coupling, bacterial glutamine transaminase-catalyzed transamination coupling, N-glycan engineering-mediated coupling, and formylglycine-generating enzyme-catalyzed reaction coupling.
(II) Technological Iteration Drives Clinical Superiority
Since the advent of antibody-drug conjugate (ADC) drugs, they can be categorized into three generations from the perspective of technological iteration.
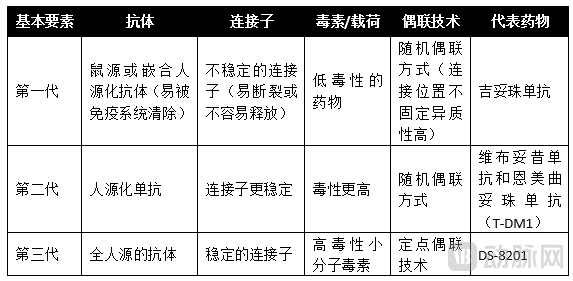
2.1 Comparison of HER2-ADCs Targeting the Same Antigen
The three key components of an antibody-drug conjugate (ADC)—the antibody, the linker, and the payload—are each critical determinants of the final drug’s safety and efficacy. Whether it is Roche’s Kadcyla (T-DM1), Daiichi Sankyo’s Enhertu (DS-8201), or RemeGen’s RC48, each has achieved an optimal balance among these three elements within its respective platform.
As a pioneer, Kadcyla opened up the field of ADC therapy for solid tumors. As latecomers, Enhertu and RC48 not only pay homage to T-DM1 but also leverage their own technological accumulation and updates to upgrade HER2 ADCs, challenging the therapeutic efficacy of T-DM1.
Target Selection:
HER2 expression in tumor cells is more than 100-fold higher than in normal cells, and the availability of mature antibody drugs targeting HER2 makes it an ideal target for antibody-drug conjugate (ADC) therapy in solid tumors. All three pharmaceutical companies have selected HER2 as the target for their ADC drug development, thereby avoiding challenges associated with unclear biological mechanisms when validating their ADC platforms.
Antibody:
T-DM1 leverages the potent endocytic activity of trastuzumab to deliver DM1 into cells, thereby exerting cytotoxic effects on tumor cells. DS-8201 also utilizes trastuzumab; its distinguishing feature is a unique linker-payload system that significantly enhances its efficacy. RC48 employs a novel, independently developed antibody, Disitamab, which targets a different HER2 epitope than trastuzumab and exhibits high selectivity for HER2. Disitamab demonstrates higher affinity, with an EC50 of 6.4 pM, compared to an EC50 of 20.1 pM for trastuzumab.
Linker:
T-DM1 utilizes the non-cleavable MCC linker, whereas DS-8201 and RC48 employ cleavable linkers (GGFG and VC, respectively). Cleavable linkers facilitate the bystander effect of antibody-drug conjugates (ADCs). Reducing the hydrophobicity of ADC drugs helps minimize their in vivo clearance and enhances therapeutic efficacy. The drug-to-antibody ratio (DAR) is also directly correlated with ADC potency. However, there is an inherent trade-off between increasing the DAR and reducing hydrophobicity; therefore, the hydrophilicity of the linker is crucial. By optimizing and screening hundreds of linker candidates, DS-8201 has established a unique linker-payload system that enables a high DAR of up to 8 while maintaining favorable hydrophilicity. This proprietary linker technology has been a key factor contributing to the success of Daiichi Sankyo’s ADCs.
Load:
Due to the limited number of antigens on the surface of tumor cells (approximately 5,000 to 10^6 antigens per cell on average) and the fact that most ADC drugs in clinical stages have an average drug-to-antibody ratio (DAR) of 3.5 to 4, only a small amount of ADC is delivered into tumor cells. This is considered one of the main reasons for the clinical failure of ADCs conjugated with conventional cytotoxic drugs such as methotrexate, paclitaxel, and anthracycline antibiotics. T-DM1 utilizes DM1, a toxin with low membrane permeability. With advances in scientific research, Daiichi Sankyo has developed DXd, a toxin with high membrane permeability, while RemeGen has adopted MMAE, a toxin with favorable properties; both exhibit higher membrane permeability than DM1. After release, these toxins can penetrate adjacent cells, producing a bystander effect. Animal studies cited in RemeGen’s prospectus revealed that the bystander effect of RC48 at 3.3 mg/kg was significantly stronger than that of T-DM1 at 10 mg/kg. Furthermore, studies have shown that the non-cleavable linker and the low membrane permeability of the DM1 toxin lead to substantial accumulation of T-DM1 in lysosomes, resulting in prolonged drug exposure, which is a significant cause of T-DM1 resistance.
2.2 The Inevitability and Contingency of ADC Success
Necessity:
Biological Certainty. The ultimate goal of cancer therapy is to selectively kill tumor cells without damaging normal cells. Chemotherapeutic agents have long remained the first-line treatment for cancer precisely due to their potent and largely non-discriminatory cytotoxic effects, although this comes with challenges of drug resistance and significant side effects. From a scientific perspective, the most straightforward rationale behind antibody-drug conjugates (ADCs) is the targeted delivery of chemotherapeutic agents or toxins even more potent than chemotherapy directly to tumor cells, leaving no room for biological or scientific doubt. If precise delivery can be achieved, success is inevitable.
Contingency:
Technological Innovation. Given that antibody-drug conjugates (ADCs) involve multiple critical factors, including antibodies, toxins, linkers, and conjugation methods, there is significant uncertainty regarding which team will achieve breakthroughs or iterative advancements in any specific area. As antibody technology continues to mature, DS-8201 has achieved a major breakthrough in targeting HER2 through the screening of toxins and linkers, as well as innovative conjugation strategies. However, it still faces the challenge of a high incidence of interstitial lung disease. In the future, new ADC products have the opportunity to achieve further iterations in drug-linker design, and even in the selection of entirely novel antibody targets.
Multinational pharmaceutical companies are heavily betting on the ADC sector, with momentum rivaling that of PD-1 in earlier years. Over the past few years, scientists worldwide have been searching for the next PD-1, but targets in the field of tumor immunotherapy, such as TIGIT, TGF-β, and biased IL-2, have all failed. So, is there a next “ADC”?
A comparison between the tumor immunotherapy (IO) sector and antibody-drug conjugates (ADCs) reveals significant differences in terms of biological certainty and technological innovation. Innovative drug development in the IO sector tends to focus more on breakthroughs in fundamental biology, such as novel targets, mechanisms, and pathways. In contrast, the primary advancements in the ADC field are currently observed in molecular design and conjugation technologies. ADCs comprise targets with well-defined mechanisms and payloads with proven efficacy, thereby presenting lower scientific risks compared to IO therapies, while imposing higher demands on technical feasibility.
Exploring druggable molecular structures from the perspective of ADC architecture, with potential extensions in the directions of ligands, linkers, and payloads:
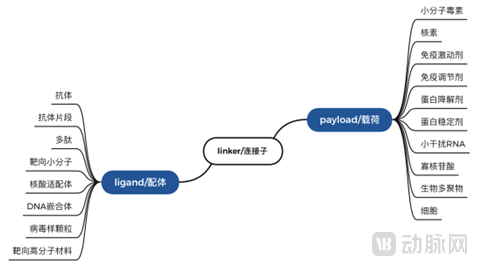
In addition to the ADC design comprising an antibody, linker, and small-molecule toxin, various permutations and combinations yield other modalities, including: Fragment-Drug Conjugates (FDC), Peptide-Drug Conjugates (PDC), Small Molecule-Drug Conjugates (SMDC), Radionuclide-Drug Conjugates (RDC), Immune-Stimulating Antibody Conjugates (ISAC), Antibody-Degrader Conjugates (ADeC), Antibody-Cell Conjugates (ACC), Virus-like Drug Conjugates (VDC), Antibody-Oligonucleotide Conjugates (AOC), and Antibody-Biopolymer Conjugates (ABC).
Classification by Ligand Type:
Antibody Fragment-Drug ConjugatesThe primary difference between (FDC) and ADC lies in replacing the full-length antibody with an antibody fragment. This approach is mainly driven by advances in novel antibody engineering technologies. The smaller antibody structure may offer superior efficacy in tumor penetration; however, the reduced size may also impose greater constraints on the selection of conjugation sites, potentially leading to differences in drug metabolism compared to full-length antibodies.
Peptide-Drug Conjugates(Peptide-Drug Conjugates, PDC) primarily involve replacing the antibody moiety in Antibody-Drug Conjugates (ADCs) with homing peptides. Compared to currently available ADC drugs, PDC drugs offer several advantages, including lower molecular weight, enhanced tumor penetration, reduced immunogenicity, scalability via solid-phase synthesis, lower production costs, and relatively favorable pharmacokinetics. However, current peptide drugs are mainly derivatives of endogenous peptides, such as octreotide, GLP-1, and insulin. Therefore, developing high-affinity targeted peptides may require more extensive research than the antibodies used in ADCs.
Small Molecule Conjugate Drugs(Small Molecule-Drug Conjugates, SMDCs) employ small molecule targeting, and site-specific conjugation techniques between small molecules can achieve a relatively precise drug-to-antibody ratio (DAR). Composed entirely of small molecules, SMDCs offer easier cost control; they are theoretically non-immunogenic, facilitating safety management; and their significantly lower molecular weight compared to Antibody-Drug Conjugates (ADCs) enables better cellular permeability in solid tumors as well as improved in vitro and in vivo stability. However, the selection of small molecule ligands for SMDCs is not as straightforward as the antibody selection for ADCs, posing greater challenges in screening suitable ligands. Additionally, while the low molecular weight offers advantages, factors such as half-life and the feasibility of oral administration must also be considered.
Broadly speaking, Proteolysis-Targeting Chimeras (PROTACs) also fall under the category of Small Molecule-Drug Conjugates (SMDCs), utilizing small molecules to targetably recruit E3 ligases to specific targets, thereby inducing degradation of the target protein. However, PROTACs require non-cleavable linkers, whereas SMDCs employ cleavable linkers. From the perspective of cleavability, small-molecule prodrug design can also be classified as SMDCs, although prodrugs do not necessarily require a targeting ligand.
Similarly, aptamer-drug conjugates (ApDCs), DNA chimera conjugates (such as DNA chimera-targeted hydrolysis, DENTAC), and virus-like particle conjugates (VDCs) primarily replace antibodies with entirely novel ligand components. In terms of screening difficulty, antibodies are perhaps the most accessible ligands for achieving target differentiation.
Classification by Load Type:
Radionuclide-Conjugated Drugs(Radionuclide Drug Conjugates, RDC) utilize the targeted localization mediated by antibodies (Antibody-radionuclide Conjugate, ARC) or small molecules (including peptides, Peptide-radionuclide Conjugate, PRC) to precisely deliver cytotoxic/imaging agents (radionuclides/radioisotopes) to target sites, thereby avoiding the potential hazards of systemic exposure. The distinction lies in the fact that the payload of RDCs is a radionuclide, enabling both diagnostic and therapeutic functions. Compositionally, RDCs also differ slightly from Antibody-Drug Conjugates (ADCs), requiring specific functional group structures to chelate the radionuclide. Representative drugs include Novartis’s 177Lu-DOTATATE and 177Lu-PSMA-617.
Antibody-Immune Stimulant Conjugates(Immune stimulating antibody conjugate, ISAC) and Immune modulating antibody conjugate (IMAC) primarily involve conjugating immune agonists or immunosuppressants to antibodies to achieve targeted release of the active ingredients. Due to the widespread distribution of their downstream effector cells in the body, if effective targeting is not achieved, it would not only limit the administrable dose and compromise efficacy but also lead to off-target toxicity due to systemic distribution. Therefore, antibody-mediated targeting represents an ideal approach. Drugs involving this mechanism mainly include BDC-1001, a Toll-like receptor (TLR) agonist-based ISAC; XMT-2056, a STING agonist-based ISAC; and ADCT-301, an IMAC drug that regulates Treg cells.
Antibody-Degrader Conjugates(Antibody Degrader Conjugate, ADeC). Its technical principle involves using protein degraders, such as molecular glues or PROTACs, as payloads, thereby combining the tumor specificity of antibodies with the applicability of PROTAC molecules at catalytic doses for low-abundance targets. A representative drug is ORM-5029, developed by Orum Therapeutics. In contrast to degraders, another drug design strategy involves protein stabilizers. DUBTACs are also bifunctional molecules similar to PROTACs; one end consists of a compound that binds to the disease-causing protein, which is linked via a linker to a compound capable of recruiting deubiquitinases (DUBs). Deubiquitinases can remove ubiquitin chains from the surface of proteins destined for degradation, thereby preventing their degradation and stabilizing protein levels. In 2022, several companies focused on target protein stabilization, such as Stablix and Vicinitas, completed early-stage financing; however, no companies related to Antibody-Stabilizer Conjugates (ASC) have emerged yet.
Antibody-Oligonucleotide Conjugates(Antibody Oligonucleotide Conjugate, AOC) and Antibody-siRNA Conjugates (AsiRC) are designed by conjugating oligonucleotides or siRNA to antibodies. Currently, RNA therapeutics face the challenge of insufficient extrahepatic targeting; achieving tissue-specific targeting via antibodies may represent a breakthrough direction for nucleic acid delivery. Compared with the toxins carried by Antibody-Drug Conjugates (ADCs), nucleic acid fragments have significantly higher molecular weights, and their negative charge imposes distinctly different requirements on drug design. The representative drug currently is AOC-1001, developed by Avidity Biosciences. As a pioneer in AOC development, Avidity’s design combines the tissue selectivity of monoclonal antibodies with the precision of oligonucleotide-based therapies, thereby overcoming barriers to oligonucleotide delivery and enabling targeting of genetic drivers of disease for the treatment of rare muscular disorders and other serious conditions. AOC-1001, the first AOC to enter clinical trials, consists of three components: a full-length monoclonal antibody targeting Transferrin Receptor 1 (TfR1), a linker, and an siRNA targeting DMPK mRNA, with myotonic dystrophy type 1 as its indication.
Antibody-Biopolymer Conjugates(Antibody-Biopolymer Conjugate, ABC) is obtained by conjugating an antibody to a biopolymer via a linker. A representative drug is KSI-301, developed by Kodiak Sciences. The company employs an 800 kDa branched phosphorylcholine polymer as the biopolymer. This polymer is hydrophilic and is site-specifically conjugated to the antibody through a non-cleavable linker, forming a hydration shell around the antibody that shields it from non-specific interactions, thereby facilitating targeted delivery. This platform is designed for VEGF targets to extend drug retention in ocular tissues, thereby enhancing efficacy and reducing injection frequency. It is currently being developed for the treatment of retinal vascular diseases and for preventing vision loss in patients with diabetic eye disease. In February 2022, Kodiak Sciences announced the top-line data from DAZZLE, the first registrational clinical trial of KSI-301 for wet age-related macular degeneration. The results showed that KSI-301 did not demonstrate non-inferiority to aflibercept in terms of visual acuity improvement, failing to meet the primary endpoint. The company attributed this failure primarily to the excessively long dosing interval specified in the study protocol.
Antibody-Cell Conjugates(Antibody-Cell Conjugate, ACC) functions similarly to CAR-T therapy and is more aligned with cell therapy, where antibodies serve to provide targeting for the cellular therapy, such as in CAR-NK and CAR-platelet therapies. However, antibody-cell conjugates are linked via chemical reactions without requiring genetic modification; thus, immune cells retain their natural activation signaling systems, with the antibodies serving solely a targeting role. A representative drug is ACE1702, developed by Acepodia.
Plot the innovative platforms derived from the aforementioned permutations and combinations on a graph with biological certainty as the x-axis and technological maturity as the y-axis, taking into account the following considerations:
1. Antibody-drug conjugates (ADCs) and antibody fragment-drug conjugates (FDCs) exhibit the highest degree of similarity among various conjugation platforms, and both have achieved a relatively high level of technological maturity with the advancement of antibody engineering;
2. Due to the greater difficulty in identifying ideal ligands compared to antibodies, there are fewer companies involved in the development of peptide-drug conjugates (PDCs), small-molecule drug conjugates (SMDCs), and aptamer-drug conjugates (ApDCs). These modalities exhibit relatively lower technological maturity, and the scientific certainty of demonstrating superiority over antibody-drug conjugates (ADCs) for the same target may be somewhat weaker. However, if suitable targets are identified, there remain opportunities for differentiated development.
3. From a technical perspective, Antibody-Immune Stimulator Conjugates (ISACs) share similar design methodologies with Antibody-Drug Conjugates (ADCs); however, the development of immune stimulators as monotherapies faces significant challenges, and the biological mechanisms of the payloads remain poorly understood.
4. Radionuclide Drug Conjugates (RDCs): Several drugs based on peptide and small-molecule targeting have already been launched, indicating a relatively high level of technological maturity and scientific certainty. However, due to stricter regulatory controls over radionuclides and the corresponding technical challenges that developers must overcome, RDCs are less widely adopted than Antibody-Drug Conjugates (ADCs).
5. Conjugated drugs in protein degradation systems include simple PROTACs, antibody-mediated ADeCs, and DNA chimera-mediated DNEtacs. All of these rely on the intracellular ubiquitin-proteasome system. While their biological mechanisms have been relatively well studied, many technical challenges remain to be overcome in drug molecular structure design and in the formation of intracellular ternary complexes during the onset of action. Currently, related PROTAC molecules have entered clinical trials, while others remain in the preclinical stage.
6. The mechanism of action of protein stabilizers is opposite to that of protein degraders; they inhibit the ubiquitin-proteasome system, acting in contrast to the body’s endogenous ubiquitination machinery. Compared with degraders, protein stabilizers require further research in both basic biology and molecular design, and currently, some companies are still in the preclinical stage.
7. Antibody-Oligonucleotide Conjugates (AOCs) address the core challenge of targeting in current nucleic acid therapeutics. Compared with Antibody-Drug Conjugates (ADCs), AOCs exhibit distinct linker technologies and areas requiring further exploration. Regarding biological certainty, although the payloads involve novel nucleic acid sequence designs, these sequences can be designed by referencing genomics data. Similar to other RNA-based drugs with high development success rates, AOCs face certain challenges in biological maturity that remain relatively controllable.
8. Virus-like drug conjugates (VDCs) and antibody-biopolymer conjugates (ABCs) involve larger drug molecules with more complex structures; their overall biological certainty remains lower, and the relevant technologies are still in earlier stages of development.
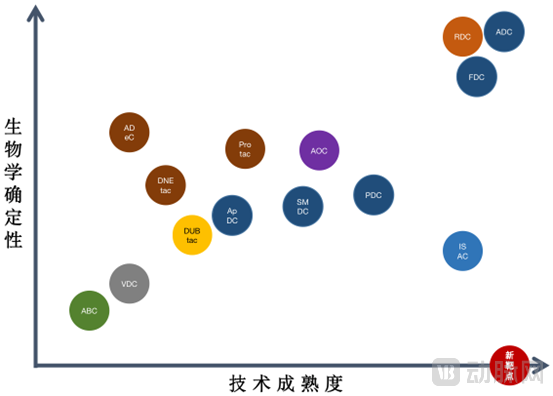
Note: 1. On the coordinate plane, ADCs are positioned as having the highest biological certainty and technological maturity, while mature molecular formats (e.g., antibodies, small molecules) for novel targets are positioned as having the lowest biological certainty but the highest technological maturity. 2. Different colored blocks correspond to technology platforms with significant differences, whereas the same color indicates that the respective platforms have good substitutability in terms of technical pathways or practical applications.
As shown in the figure above, in addition to the relatively mature ADCs, FDCs, and RDCs, fields poised for breakthrough advancements with the improvement of conjugation technologies and understanding of molecular types include protein degradation conjugates and antibody-nucleotide conjugates. Of course, beyond scientific considerations, market conditions and competitive advantages must also be taken into account; nevertheless, there remains significant potential for the emergence of next-generation “ADCs.”
References:
1.A Payday For Payloads: The Transactional Landscape Of ADCs.
2.Site-selective modification strategies in antibody–drug conjugates.
3.Cleavable linkers in antibody–drug conjugates.
4.Strategies and challenges for the next generation of antibody-drug conjugates.
5. Antibody–Drug Conjugates for Cancer Therapy.
6.Antibody-drug conjugates: recent advances in conjugation and linker chemistries.
7.Characterization of antibody-drug conjugates by mass spectrometry: advances and future trends.
8.Antibody–drug conjugates in solid tumors: a look into novel targets.
9. RemeGen Biopharmaceuticals Prospectus.
10. Iterative Advancements in HER2-ADCs.
11.Antibody–Drug Conjugates: A Comprehensive Review.
12.Antibody–Drug Conjugates (ADCs) for Personalized Treatment of Solid Tumors: A Review.
13.Collaboration on trastuzumab deruxtecan Investor conference call presentation.
14. Official websites of Avidity, Stablix, Vicinitas, Kodiak, and Acepodia.