Riding the Wave of Pharma Globalization: The $10B eCOA Market Set for Liftoff
Whether seeking direct capital injection from investment institutions or leveraging license-out deals as a means of survival, going global has become an imperative for domestic innovative pharmaceutical companies to stay afloat.
Nevertheless, the pioneers venturing overseas face an uncertain future, with water sellers along the way always... This year, Healthcare IT released a research report that surveyed the digital clinical trials subsector, specifically electronic Clinical Outcome Assessment (eCOA) solutions. The global eCOA market size was $1.47 billion in 2023 and is projected to reach $4.74 billion by 2031, representing a high CAGR of 15.80%.
Although the report did not disclose specific figures for growth in the Chinese market, the surge in overseas expansion enthusiasm seen in 2023 indicates that the annual growth rate of the domestic market has continued to accelerate.
In recent years, the FDA has been encouraging pharmaceutical companies to adopt Digital Health Technologies (DHTs) in their applications. Currently, approximately 80% of clinical trials in the United States are conducted using electronic Clinical Outcome Assessments (eCOA), with 60% of those in Europe following the same approach.
In China, fewer than 5% of sponsors have adopted eCOA, while the remaining 95% of trials still rely on paper-based methods.
Seizing this opportunity, domestic and international digital clinical trial vendors have accelerated their expansion into the Chinese market while continuously innovating their service offerings. They are no longer limited to digitizing paper-based forms; instead, they are integrating technologies such as deep learning and generative AI into their solutions, embedding them deeply within the sponsorship processes for innovative drugs and even providing intuitive strategic guidance for global market entry.
Under this trend, these companies are playing an increasingly important role in the global expansion of innovative drugs, and are even poised to become an indispensable force in clinical trials for such therapies.
From project initiation to regulatory approval for market launch, an innovative drug must undergo numerous stages, including preclinical research, clinical trials, registration and approval, and post-market surveillance, with each stage carrying the risk of failure. Compounded by the unfamiliar environment encountered when expanding overseas, this process becomes exceptionally perilous.
withThe clinical study protocol is designed as follows:cases. Due to the specificities in healthcare capabilities and patient distribution across different countries or regions, the same condition may present vastly different unmet clinical needs domestically versus internationally, leading to significant disparities between domestic and overseas standard treatments. For instance, for certain types of malignant lymphoma, the standard treatment in China may be conventional chemotherapy, whereas in the United States and Europe, there is a greater preference for already-approved targeted therapies.
Therefore, accurately identifying the unmet clinical needs of overseas populations is a critical consideration for domestic sponsors. After all, some pharmaceutical companies have previously failed to clearly define the future standard-of-care treatments they aimed to benchmark against, nor did they establish clear plans for efficacy and safety. Only at the final stage did they discover fundamental flaws in their initial protocols, rendering their candidates unable to pass clinical trials. This resulted in the loss of tens of millions of yuan in investment and several years of effort.
Differences in Research PlansThis also constrains the global expansion of innovative pharmaceutical companies. As the United States is an immigrant nation, there is a discrepancy between the FDA’s requirements for subject enrollment numbers and racial distribution in clinical trials and those of the NMPA. Consequently, sponsors must take racial demographics into account when selecting trial sites.
In reality, many domestic sponsors are unfamiliar with the actual conditions of overseas trial sites. They often slow down their submission processes due to uncertainties regarding patient distribution at local trial centers and slow recruitment rates. Furthermore, the lack of real-world data to support enrollment plans makes it difficult to identify high-performing research centers, which can also lead to enrollment falling short of expectations during the recruitment phase.
Research Implementation ProcessThere is also a more straightforward issue. Due to differences in national contexts, the quality management methods and scale formats adopted may vary to some extent. Many domestic pharmaceutical companies are accustomed to 100% Source Data Verification (SDV), where Clinical Research Associates (CRAs) verify whether paper-based data match the data entered into the system. In contrast, Europe and the United States have fully adopted Risk-Based Quality Management (RBQM), focusing on the review of critical data and collecting data in the form of electronic Patient-Reported Outcomes (ePRO).
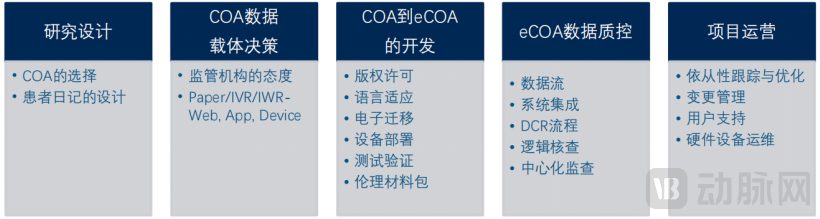
Key Stages and Essential Considerations for Implementing eCOA in Clinical Trials
Although the founding teams of many innovative pharmaceutical companies in China possess extensive experience in R&D and pipeline business development (BD) at multinational corporations (MNCs), ensuring that their teams can effectively manage every stage of overseas regulatory submissions still entails substantial trial-and-error costs. These costs represent a significant opportunity for digital clinical trial enterprises.
Products in the digital clinical trials market can be broadly categorized into two types: one comprises basic clinical trial information systems, while the other includes innovative products derived from clinical trial-related databases. Amid the current wave of Chinese innovative drugs expanding into global markets, the market size for both product categories has experienced varying degrees of growth.
The clinical trial information system market has long been highly concentrated and, at one point, approached saturation. In 2021, the top six vendors, including Taimei Medical Technology and Veeva, accounted for more than two-thirds of the market share. The adoption rates of products such as EDC, RTSM, and PV at trial sites had already reached 90%–100%, achieving near-complete market coverage. Consequently, incremental growth driven by overseas expansion is primarily derived from niche segments such as eCOA.
Compared with products such as EDC and PV, eCOA focuses on the assessment of clinical outcomes, and its priority in China’s pharmaceutical industry is lower than that of data collection and compliance. Meanwhile, the initial implementation cost of eCOA can be relatively high, including expenses for system acquisition, personnel training, and data migration. Before seeing a clear return on investment, many innovative pharmaceutical companies remain cautious about adopting this system.
But now, going global has become an inevitability. The former disadvantages of eCOA have become new breakthrough points for companies specializing in the digitalization of clinical trials. In addition to multinational corporations like Medidata and Veeva accelerating their efforts, domestic players such as Taimei, Bio-Knowing, and Tigermed are increasing their investments, while startups like Yaosheng Health Technology, Quanrong, and Yilin Cloud are also entering the market.
Revisiting Innovative Products in the Digital Clinical Trials Market. Based on actual FDA submission experiences, configuring eCOA (electronic Clinical Outcome Assessment) is relatively straightforward to accommodate. However, to address clinical protocols, clinical plans, and even post-marketing drug marketing challenges, providers of digital clinical trial services must possess extensive overseas operational experience to effectively navigate “localization” issues when expanding globally.
Compared with domestic enterprises, multinational corporations hold a slight advantage due to their experience and background, and are also expanding the market size of clinical trial digitalization through product innovation.
For instance, when facing the aforementionedUnmet Clinical Needs, the medical affairs departments of most domestic innovative pharmaceutical companies determine patient needs primarily based on published literature; however, such literature is largely written around drugs already on the market, and the study populations are typically specific groups that align with the drugs’ characteristics.
This means that pharmaceutical companies, when formulating early-stage research strategies, can only rely on limited published literature to make critical decisions regarding clinical trials. As a result, they struggle to identify specific patient populations that have not been adequately studied and are reluctant to invest tens or even hundreds of millions in these data-unvalidated areas. Ultimately, many new drugs fail at the final stage due to insufficient innovation.
In response to such needs, some companies have already developed innovative solutions. Among them, Medidata has attempted to leverage historical clinical trial data to help Fast Followers identify patient populations that differentiate them from competitors, thereby assisting pharmaceutical companies in better selecting unmet clinical needs and assessing project feasibility.
According to Medidata, they previously collaborated with a biopharmaceutical company attempting to develop drugs targeting KRAS mutations. Given the scarcity of published literature on this target, the company sought to expand its understanding of the KRAS-mutant population and identify unmet medical needs and complications among this target population under standard-of-care (SoC) chemotherapy regimens.
During implementation, Medidata established a dataset comprising over 800 patients with KRAS mutations and employed specific methodologies to test the selected hypotheses proposed by the client. Within weeks, Medidata identified areas of unmet therapeutic need, such as unexpected complications including brain metastases, thereby ultimately pinpointing a patient population differentiated from that of competitors.
Regarding the research plan, the competition here mainly stems fromPatient RecruitmentThe United States is a nation of immigrants, and it places particular emphasis on racial demographics in clinical trials, with specific requirements for the proportions of White, Black, and Hispanic participants. Chinese sponsors are also expected to recruit patients according to similar demographic ratios.
Meeting such proportional requirements is inherently challenging, and fierce competition for patient recruitment is further constraining the conduct of clinical trials. Although the United States accounts for only 4.25% of the global population, it bears approximately 35% of the global demand for patients in clinical trials. In recent years, the FDA has faced a growing number of submissions, while patient availability remains insufficient. As a result, 50% of clinical trial sites have failed to enroll patients according to their original plans, leading to significant increases in both the duration and costs of pharmaceutical companies’ trials, and even causing the abandonment of numerous clinical trials.
To address this issue, many overseas third-party research organizations have launched site selection services to meet sponsors’ needs for global clinical trial site identification. These solutions can be used for site screening and prediction, identifying suitable sites based on the sponsor’s trial characteristics and contextual factors. They even leverage data-driven predictions to ensure trials proceed as planned and enable real-time adjustments to trial forecasts as new data become available.
The overseas expansion strategies of China’s leading enterprises have recently begun to yield significant results. As early as 2021, when Taimei submitted its prospectus, its TrialOS platform for collaborative drug R&D had already integrated with regulatory databases across 34 countries and regions. Tigermed has gone even further, establishing overseas subsidiaries in 10 countries across the Asia-Pacific, North America, and Europe, directly targeting international clients.
Whether it is the deep learning of the past or the large language models that are currently sweeping across the industry, every clinical trial digitalization enterprise has shown great enthusiasm for this innovative technology. In the wave of global expansion, differing perceptions of AI technology have also endowed them with varying competitive advantages.
If digital clinical trial enterprises are further segmented based on their AI capabilities, the two industry leaders, Taimei and Veeva, undoubtedly fall into the same category. Specifically, both companies are among the few in the industry capable of providing digital solutions that span from clinical development to commercialization, and both have integrated AI across their entire product portfolios.
From Veeva’s perspective, AI can enhance data processing efficiency, optimize patient recruitment, improve trial design, and increase the precision of data analysis, thereby reducing clinical trial costs and accelerating regulatory submission processes. This enables Veeva to provide stronger core capabilities when serving Chinese clients expanding into global markets.
Medidata can be categorized as another type of enterprise. In addition to applying AI within its workflows, it has also pioneered numerous innovative AI-driven applications.
In 2020, the FDA approved the use of Medidata’s AI Synthetic Control Arm (SCA) solution in a Phase III registrational trial of MDNA55 for recurrent glioblastoma (rGBM) conducted by Medicenna Therapeutics, a U.S.-based clinical-stage immunotherapy company. This marks the first approval of a hybrid external control arm in a Phase III trial and represents a typical case of AI “data application.”
Interleukin-4 receptor protein therapeutics are the main component of MDNA55, which can induce cancer cells to absorb and take up the drug, leading to the release of toxins and resulting in immunogenic cell death. However, due to challenges such as difficulties in recruiting and retaining trial participants, the trial once faced a dilemma of having no available subjects.
On the one hand, as a targeted therapeutic macromolecule, MDNA55 cannot cross the blood-brain barrier and must be delivered directly to the tumor during administration, making it impossible to conduct controlled trials with a placebo-recruited control group. On the other hand, patients with recurrent glioblastoma have limited survival times, and many who urgently need treatment decline to participate in clinical trials due to concerns about being assigned to the control group.
To address Medicenna’s needs, Medidata first designed a hybrid control arm protocol for the Phase III trial based on the results of the MDNA55 Phase II trial. It then leveraged its own database of more than 7 million anonymized patient records to construct a synthetic control arm that matched the patient characteristics. Finally, Medidata employed propensity score matching, a statistical method, to ensure that the AI-enabled synthetic control arm was well-balanced and comparable to the treatment arm.
Medidata successfully helped Medicenna reduce the enrollment of 100 control-group patients in its upcoming Phase III trial, marking a pioneering use of hybrid external controls in rare disease trials. This application of AI-generated synthetic control arms will increase the probability that enrolled patients receive the investigational drug, is expected to improve enrollment rates, and accelerate trial timelines without compromising the scientific interpretability of the study.
Overall, whether through quality improvement and efficiency enhancement across the entire workflow or by creating AI-generated synthetic control groups for specific scenarios, the integration of AI has enabled digital clinical trial companies to engage more deeply in new drug clinical trials, thereby more effectively facilitating the market launch of innovative drugs.
Meanwhile, innovative pharmaceutical companies’ global expansion will also benefit from continuous breakthroughs in AI technology. Empowered by more efficient digital tools for clinical trials, we may see more high-quality domestic products gain regulatory approval at an accelerated pace, thereby benefiting patients worldwide.