China's New Regulation on Biomedical Novel Technologies Establishes Dual-Track Pathway for Clinical Translation with Streamlined Access, Oversight, and Controlled Reimbursement
On October 10, 2025, the full text of the “Regulations on the Clinical Research and Clinical Translational Application Management of New Biomedical Technologies” (hereinafter referred to as the “Regulations”) was unveiled.
Nearly a month ago, on September 12, 2025, the State Council’s executive meeting reviewed and approved the “Regulations on the Administration of Clinical Research and Clinical Translational Application of New Biomedical Technologies (Draft).” The meeting pointed out that vigorous efforts should be made to promote technological innovation and development in China’s biomedical sector, accelerate technology research and development as well as the transformation of achievements, facilitate quality improvement and upgrading of the biopharmaceutical industry, and focus on cultivating new competitive advantages for development.
China’s regulatory exploration of novel biomedical technologies is far from a blank slate. From the 2015 trial implementation of the Administrative Measures for Clinical Research on Stem Cells (Trial) to the drafting of the 2019 Regulatory Regulations on the Clinical Application of Novel Biomedical Technologies (Draft for Comments) (hereinafter referred to as the “Draft Regulations”), the regulatory framework has gradually become clearer through years of practice. However, as China ventures into the “no-man’s-land” of global innovation in certain fields, issues such as inconsistent standards, ambiguous translation pathways, and coexisting ethical controversies and technical risks have become increasingly prominent.
In this context, high expectations have been placed on the introduction of the Regulations. It is widely believed within the industry that regulating clinical research and translational applications will not only help promote medical progress but also safeguard the quality and safety of healthcare, thereby upholding human dignity and protecting life and health.
“The Regulations” came into effect on May 1, 2026, with one of the most significant changes being a major shift from pre-approval to filing-based management for clinical research.In the future, clinical studies can be initiated once they have passed internal academic and ethical reviews at the institution and completed registration on the national platform. This means that the “starting gun” for research is now in the hands of medical institutions more familiar with frontline scientific research, while the role of national regulation has shifted from “gatekeeper” to “referee,” focusing on strengthening oversight of the research process and penalizing violations. This achieves tighter process regulation while loosening access controls.
In the stage of achievement transformation, the Regulations have significantly streamlined the approval process. More importantly, they have established a “dual-track” system for clinical translation. Upon completion of clinical studies, new technologies may either follow the traditional registration pathway or apply for clinical application via the technical pathway under the Regulations. This reform provides a more flexible and faster route to market for frontier technologies, particularly cell therapy products.
However, deregulation does not mean laissez-faire. In tandem with the loosening of restrictions, the Regulations establish stringent penalties, including substantial fines and even lifetime bans from practicing, while reinforcing requirements for long-term data retention and subject follow-up.
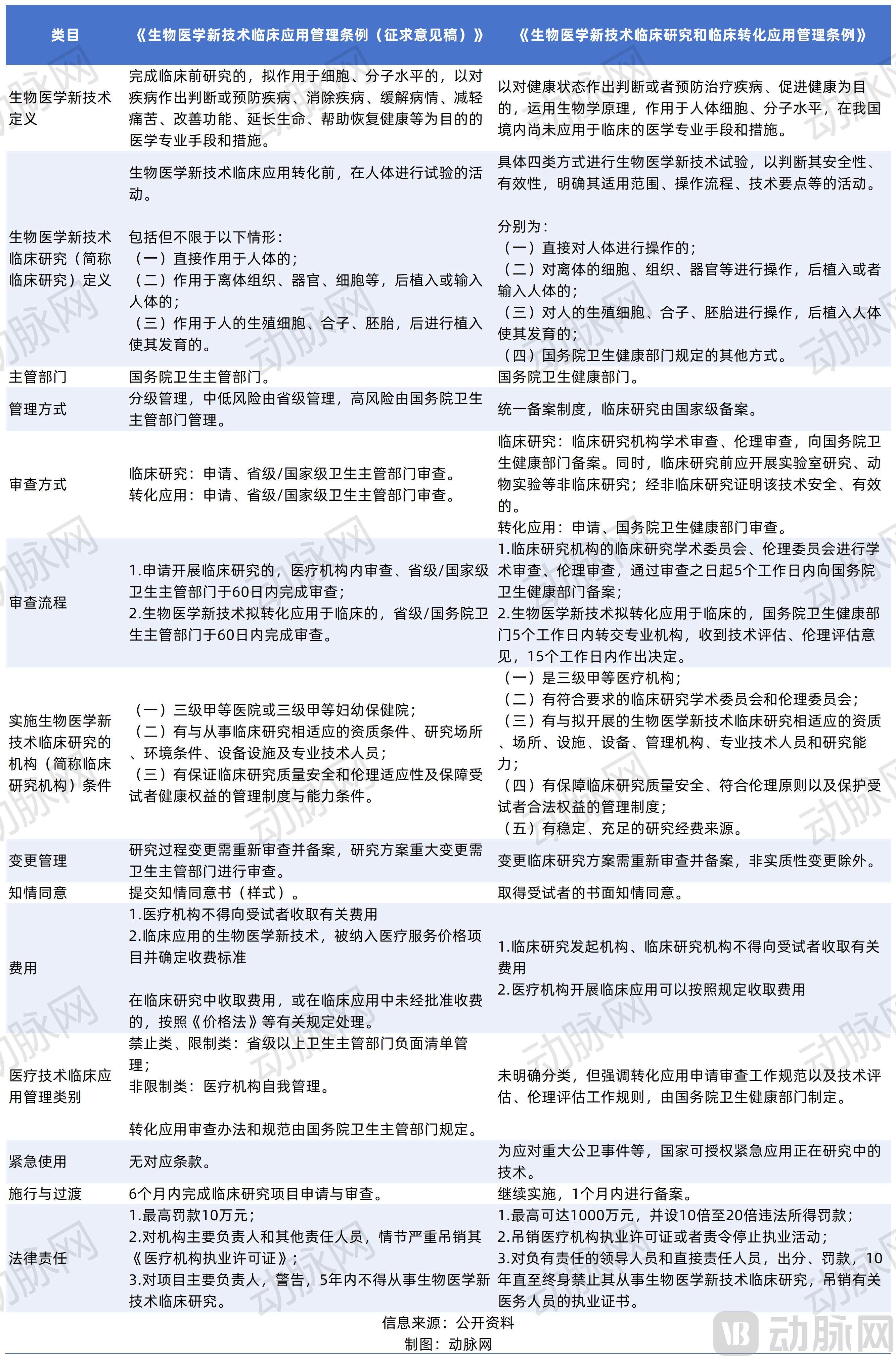
《Regulations on the Clinical Application and Management of New Biomedical Technologies (Draft for Comments)》Overview Chart Comparing the Draft) and the Regulations on the Clinical Research and Clinical Translation Application Management of New Biomedical Technologies
Strengthen the Regulatory Framework and Define Market Entry Criteria
In fact, China has accumulated years of experience in the management of clinical research on new biomedical technologies. As early as 2015, the former National Health and Family Planning Commission and the former China Food and Drug Administration jointly issued the Administrative Measures for Clinical Research on Stem Cells (Trial), establishing a management model for stem cell clinical research with medical institutions as the responsible entities. After ten years of practice, these regulations have effectively promoted the development of stem cell research and the improvement of product quality in China, laying a solid foundation for the promulgation of the Regulations.
In February 2019, the National Health Commission issued a Draft Opinion, proposing for the first time the implementation of tiered management for clinical research involving new biomedical technologies. According to the Draft Opinion, medium- and low-risk technologies are to be regulated by provincial health authorities, while high-risk technologies fall under the jurisdiction of the national health authority under the State Council. High-risk technologies encompass several frontier fields, including gene editing, stem cell technology, application of xenogeneic biomaterials, and assisted reproduction.
Shen Jianzhong, Deputy Director of the China National Center for Biotechnology Development, pointed out that there are still two major challenges in the practical development of new biomedical technologies: first, ethical controversies and technical risks associated with new technologies can easily trigger public skepticism; second, some scientific research achievements fail to truly benefit patients, as the pathway for translating certain new technologies from research to clinical practice remains unclear.
Meanwhile, China has reached the global frontier of technological innovation in certain emerging biomedical fields, venturing into uncharted territory. Industry experts note that, taking the stem cell sector as an example, the lack of unified standards hinders mutual recognition of research data, directly impacting the review and approval process. Wu Zhaohui, Vice Chairman of the Chinese Medicinal Biotech Association, emphasized, “During the initial phase of implementing the filing system for clinical research on stem cells, there were indeed significant issues with the quality of many preparations.”
Against this backdrop, a robust regulatory framework and legal safeguards can better support clinical research and the translational application of new biomedical technologies.
Recently, officials from the Ministry of Justice and the National Health Commission answered reporters’ questions on matters related to the Regulations.The Regulations apply to all clinical research, clinical translation and application, and related regulatory activities involving new biomedical technologies conducted within the territory of China. Meanwhile, the health administrative department of the State Council, in conjunction with the drug regulatory department, shall formulate and, as appropriate, adjust the guiding principles for delineating the boundaries between new biomedical technologies and pharmaceuticals or medical devices, in light of scientific and technological developments.
Gao Jianchao, an Associate Researcher at Changping National Laboratory and a former senior review expert at the Center for Drug Evaluation (CDE), pointed out from a regulatory perspective that stem cell clinical research in China exhibits a structural characteristic of “many early-stage studies but few late-stage ones.” He particularly emphasized that while fully leveraging the advantages of China’s unique Investigator-Initiated Trial (IIT) mechanism, it is essential to prioritize data integrity and compliance to lay a solid foundation for subsequent drug applications.
It took nearly six years from the release of the “Draft Opinion” to the formal implementation of the “Regulations.”
Compared with the previously released Draft for Comments, the Regulations feature a more streamlined structure, adjusted from 7 chapters and 63 articles to 7 chapters and 58 articles, with optimizations made in several key areas.
In the Draft Opinion, new biomedical technologies are defined as “medical professional methods and measures that have completed preclinical research, are intended to act at the cellular or molecular level, and aim to diagnose diseases, prevent diseases, eliminate diseases, alleviate disease conditions, relieve pain, improve functions, prolong life, and facilitate recovery of health.” Whereas“The Regulations” further define new biomedical technologies as medical professional methods and measures that “act at the cellular or molecular level of the human body and have not yet been applied clinically within China.”
The Regulations further state that clinical research on new biomedical technologies refers to the experimental application of such technologies through the following methods: (1) direct manipulation of the human body; (2) manipulation of ex vivo cells, tissues, or organs, followed by implantation into or infusion into the human body; (3) manipulation of human germ cells, zygotes, or embryos, followed by implantation into the human body to enable development; and (4) other methods prescribed by the health administrative department of the State Council. Under this definition, various cell therapies, gene therapies, xenotransplantation, and similar interventions are included.
According to the Regulations, the clinical study sponsor and the research institution shall enter into a written agreement to clarify their respective rights and responsibilities and jointly develop the study protocol. Clinical research institutions may also initiate studies independently, thereby further broadening the pathways for participation by research entities.
Regarding institutional access,The Regulations specify that clinical research institutions must be Grade III, Class A medical institutions, and must have qualified academic committees and ethics committees, as well as stable and sufficient research funding support.
“Filing + Approval” Management, Clarifying Compliant Fee Charging
In terms of management model, the Regulations have achieved a significant shift from pre-approval to record-filing management for clinical research.
The Regulations clarify the fundamental prerequisites and prohibited red lines for clinical research, stipulating that new biomedical technologies may only be used in clinical research after their safety and efficacy have been demonstrated through non-clinical studies and after they have passed academic and ethical reviews. Technologies explicitly prohibited by laws and administrative regulations, or those posing significant ethical concerns, shall not be subject to clinical research. Furthermore, the Regulations support the conduct of clinical research under a filing-based management system, specify the conditions that clinical research institutions must meet, detail the filing procedures, and require the health department of the State Council to assess filed projects, promptly correcting or halting them if risks are identified.
In other words,The authority to initiate clinical studies has been delegated to the institutions themselves. Studies may commence once they have passed the institution’s internal academic and ethical reviews and completed national filing within five working days.The role of national regulatory authorities has shifted from “market access gatekeeping” to “process oversight,” with a heightened focus on the review of filing information, supervision of the research process, and enforcement against non-compliant practices.
In the approval phase for translational application following the completion of clinical studies, the Draft Opinion specifies that such applications must undergo review by health administrative authorities at two levels. The entire process theoretically requires a minimum of 120 days, excluding time required for document circulation and other logistical procedures.
"The Regulations" significantly streamline the approval process,Clinical study sponsors shall submit applications directly to the health department under the State Council. The health department under the State Council shall forward the applications to professional institutions within five working days, and make a decision within 15 working days upon receipt of the technical and ethical assessment opinions. The statutory time limit for the entire process is reduced to approximately 20 working days.From this perspective, the Regulations have eliminated the provincial-level preliminary review process, thereby avoiding potential issues of duplicate reviews and inconsistent local standards, and achieving unified and efficient approval at the national level.
In the Draft Opinion, clinical application management of medical technologies is categorized into prohibited, restricted, and unrestricted classes. Prohibited and restricted medical technologies are subject to negative list management and strictly regulated by health authorities at or above the provincial level; unrestricted medical technologies are self-managed by medical institutions. The review measures and standards for translational application shall be stipulated by the health authority under the State Council. Although the Regulations do not explicitly specify such classification, they emphasizeThe specifications for the review of applications for translational use, as well as the rules for technical and ethical assessments, shall be formulated by the health administrative department of the State Council.
Furthermore, the Regulations strengthen the management of the clinical research implementation process, requiring clinical research institutions to strictly adhere to the filed protocol, implement risk prevention and control measures, and conduct follow-up monitoring of subjects after the study concludes to assess the long-term safety and efficacy of the technology. Taking data management and long-term follow-up as examples, the Regulations require that clinical research records and source materials be retained for 30 years from the end of the study; studies involving offspring must have their data preserved permanently, thereby enhancing the traceability of research data and long-term accountability. Clinical research institutions shall conduct follow-up monitoring of subjects to evaluate the long-term safety and efficacy of new biomedical technologies.
Meanwhile, the Regulations impose stringent legal liabilities.In addition to hefty fines, the regulations stipulate that responsible individuals may be subject to industry bans ranging from 10 years to a lifetime, and medical personnel shall have their practicing certificates revoked.。
The Regulations also establish an emergency use mechanism. For applications seeking clinical translation and application of new biomedical technologies to treat life-threatening conditions with no effective treatments available, or to address urgent public health needs, the health administrative department of the State Council shall prioritize their review and approval. In response to particularly major public health emergencies or other urgent events that seriously threaten public health, the health administrative department of the State Council may, upon organized demonstration confirming necessity, authorize the emergency use of new biomedical technologies currently undergoing clinical research, within a specified scope and timeframe.
Notably, the Draft Opinion explicitly stipulates that medical institutions shall not charge participants any fees related to the research content in any form. After approval for clinical application, the relevant items shall be included in the medical service price catalog and the charging standards shall be determined by the provincial-level government’s medical pricing authorities in conjunction with the health authorities. WhereasThe Regulations stipulate that, upon completion of clinical research on new biomedical technologies, an application for translational application may be submitted to the health administrative department of the State Council; medical institutions approved to carry out clinical applications may charge fees in accordance with relevant provisions.
“This directly accelerates the clinical implementation and promotion of new biomedical technologies,” said Gao Yi, Director of the Translational Medicine Center at Zhujiang Hospital of Southern Medical University and Director of the Guangdong Provincial Engineering Technology Research Center for Artificial Organs and Tissue Engineering. “In the past, there was only one pathway—market approval of new drugs or medical devices—but now an additional avenue has emerged.” With policy encouragement,New technologies will be gradually introduced into clinical practice and, for a certain period, may exist in the form of “in-house hospital preparations.”
Some investors also believe that investigator-initiated trials (IITs) under the dual-track system are a significant source for the early identification of differentiated projects. Early-stage translational projects within this dual-track framework require clearer pipeline planning and higher standards for team capabilities.
Previously, the transition of stem cell therapy from clinical research to clinical application followed a dual-track system for advanced therapies, namely two regulatory pathways: medical technology and drug registration.
The dual-track system has been further streamlined and standardized. In the future, products such as cell therapies may choose to undergo registration and approval via the pharmaceutical pathway, or pursue clinical translation through the technical pathway stipulated in the Regulations.
It is widely recognized within the industry that under the dual-track system, quality control must be strengthened to ensure the authenticity and reliability of data. As early as when the “Administrative Regulations on Clinical Research and Clinical Translation Application of New Biomedical Technologies (Draft)” were reviewed and approved, Zhang Yu, Deputy General Manager and Chief Scientific Officer of Vcanbio, had already stated that the industry needs to proceed with caution amidst high enthusiasm.
After six years of meticulous development, the Regulations have, to a certain extent, filled the regulatory gap for new biomedical technologies in China, marking a transition toward a more mature and scientific regulatory framework.