PERT: A Disease-Agnostic Gene Editing Platform Targeting Nonsense Mutations for the Treatment of Thousands of Genetic Disorders
Globally, more than 8,000 genetic diseases collectively affect hundreds of millions of patients, yet existing gene-editing therapies face a fundamental challenge:Each disease, and even each mutation, requires the development of a distinct treatment regimen.
Although more than 70 gene-editing clinical trials have demonstrated efficacy in diseases such as sickle cell disease and hypercholesterolemia, it remains impractical from regulatory, development, and manufacturing cost perspectives to develop independent therapeutic regimens for each of the more than 200,000 known pathogenic mutations.
A groundbreaking study published in the journal Nature on November 19, 2025, has brought new hope to this predicament. Researchers from the Broad Institute and Harvard UniversityDavid Liu's TeamA method namedPERT(Prime editing-mediated readthrough of premature termination codons) innovative strategy, achieving disease-agnostic gene editing for the first time—A single tool can treat multiple genetic diseases caused by the same type of mutation.

(Source: Nature)
As Liu stated in an interview with Nature, “Disease-agnostic approaches like this bring incredibly exciting possibilities for patients.” This study not only represents a major technological breakthrough but, more importantly, has the potential to fundamentally transform the treatment paradigm for genetic diseases.
In the human genome, DNA sequences encode protein information in the form of codons. However, when a gene mutation converts a codon that originally encoded an amino acid into a stop codon (TAG, TAA, or TGA), it results in a nonsense mutation, causing the cellular protein synthesis machinery to prematurely terminate translation and produce truncated, typically nonfunctional proteins.
According to statistics from the ClinVar database,Nonsense mutations account for 24% of known pathogenic alleles and are one of the leading causes of genetic diseases.Although these nonsense mutations are distributed across different genes and cause distinct diseases, they share a common molecular consequence: premature termination of translation before a full-length functional protein can be produced. This shared mechanism provides a theoretical foundation for the development of universal therapeutic approaches.
Suppressor tRNA (sup-tRNA) is a special class of transfer RNA molecules whose anticodons can recognize stop codons and insert an amino acid at these positions, thereby allowing protein synthesis to continue and read through sites that should have terminated translation. This mechanism provides an elegant solution for treating nonsense mutations.
However, a reasonable concern regarding sup-tRNA is whether it might also read through normal stop codons at the ends of genes, thereby producing abnormally elongated proteins.
Multiple studies have shown that this risk is actually low, for reasons including: the distribution pattern of premature termination codons differs from that of natural stop codons; multiple redundant stop codons often follow natural stop codons; and various mechanisms within cells prioritize the recognition of natural stop codons. Therefore, in eukaryotes, both naturally occurring and exogenous sup-tRNAs can be expressed without significant toxicity.
Although the therapeutic potential of sup-tRNA has long been recognized, its clinical application still faces significant challenges. Both viral vector delivery and lipid nanoparticle delivery carry the risk of triggering immune responses and impose the burden of repeated dosing. Even when delivery is successful, existing methods often require high-level overexpression of sup-tRNA to achieve therapeutic effects, which may alter global translation and increase the risk of toxicity.
It is against this backdrop that David Liu’s team proposed a bold hypothesis:Can gene editing technology be used to permanently integrate sup-tRNA into the genome?? Enable its expression at endogenous levels to avoid repeated dosing and reduce the risk of overexpression?
A few years ago, during the “Science Karaoke” event at the David Liu Lab’s annual retreat, David Liu proposed the idea of combining suppressor tRNAs with prime editing technology.
“Scientific Karaoke,” despite its name, does not involve singing; rather, it is a traditional activity designed to encourage team members to propose bold and imaginative project ideas. Each member spends 10 minutes presenting a breakthrough concept that could steer the laboratory in a new direction. At the end of the presentations, David Liu asked whether any lab members were willing to take on the proposed project, and several volunteered, thereby launching the PERT project.
The core of the PERT strategy isPrime Editing Technology, this is a precise gene-editing tool previously developed by Liu’s team. Unlike traditional CRISPR systems, prime editors combine a programmable nickase with a reverse transcriptase, enabling them to search, cut, and replace target DNA sequences with new ones—much like a word processor—without causing double-strand breaks, thereby offering greater safety and precision.
The ingenuity of the PERT strategy lies in its use of Prime editing to permanently convert one of the 418 endogenous tRNAs in the human genome into an optimized suppressor tRNA through a few base changes. In this way, the engineered tRNA gene is expressed normally under its original regulatory mechanisms, without requiring the continuous presence of exogenous vectors or causing overexpression.
To realize this vision, a key question remains to be answered: Among the 418 human tRNAs, which one is most suitable for engineering into a highly efficient suppressor tRNA?
David Liu’s team conducted an unprecedented large-scale systematic screen, evaluating tens of thousands of variants across all 418 high-confidence human tRNAs. They performed iterative screens for each of the three stop codons (TAG, TGA, and TAA), including initial lentiviral sup-tRNA screening, optimization of leader and terminator sequences, saturation mutagenesis screening of individual sup-tRNAs, and saturation mutagenesis screening of specific tRNA families (such as the Leu-TAA family).
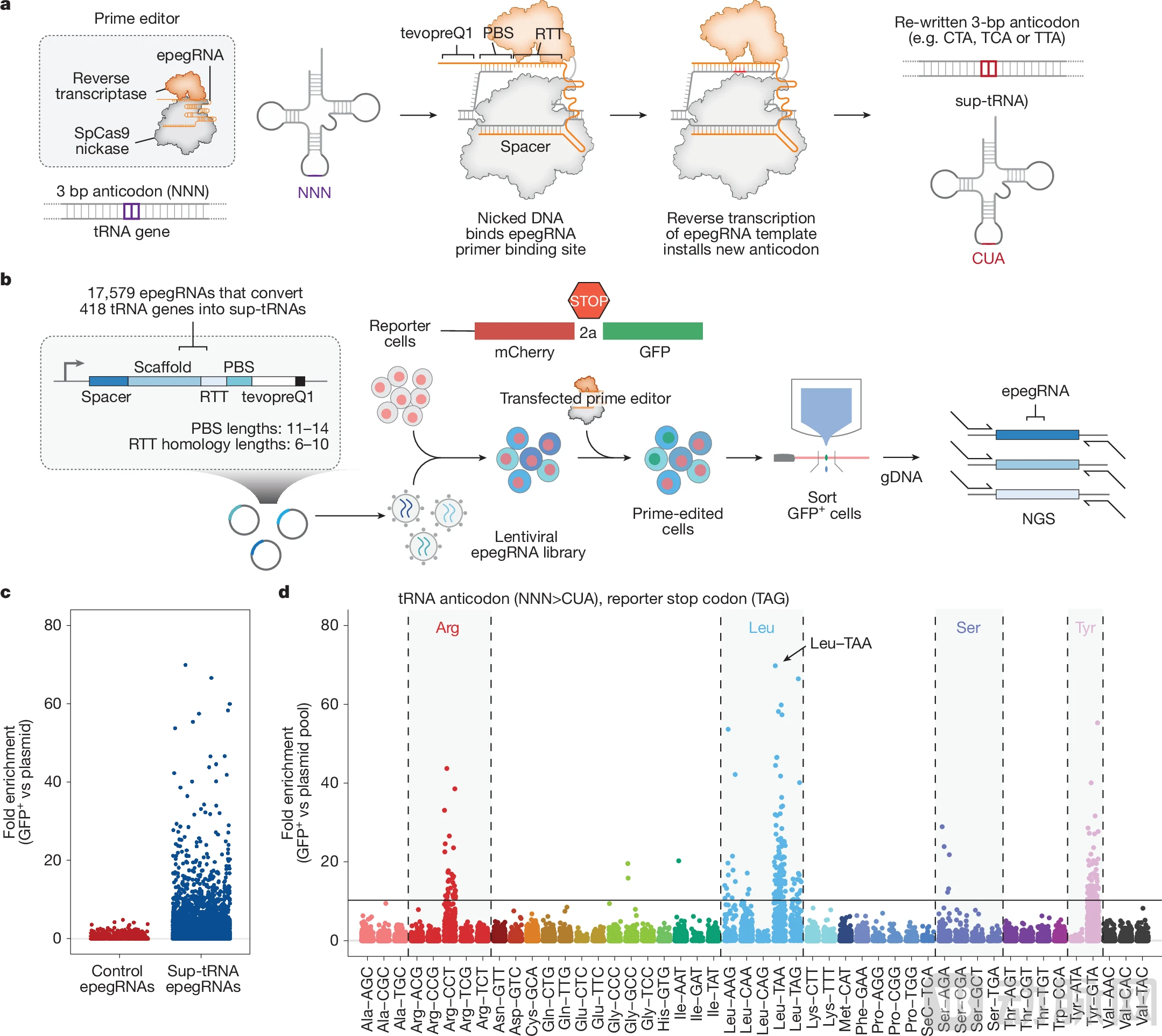
Figure: Endogenous tRNAs in mammalian cells are converted into sup-tRNAs via prime editing (Source: Nature)
Through this series of screenings, the research team ultimately identified candidate genes with the greatest potential as inhibitory tRNAs and significantly improved their readthrough efficiency through engineering optimization. Crucially, these optimized sup-tRNAs exert therapeutic effects at endogenous expression levels, without the need for overexpression.
To efficiently convert selected tRNAs into sup-tRNAs, the research team also optimized Prime editing tools by developing engineered pegRNAs (epegRNAs). These optimizations ensured precise installation of engineered sup-tRNAs at single genomic loci while preserving endogenous expression regulation.
The research team first conducted validation in human cells cultured in the laboratory. These cells carried nonsense mutations that cause four severe genetic disorders, including cystic fibrosis (a hereditary disease affecting the lungs and digestive system), Batten disease (a fatal neurodegenerative disorder), Tay-Sachs disease (a condition causing progressive damage to the nervous system), and Niemann-Pick disease (a lipid metabolism disorder).
In all of these cell models,PERT can successfully restore a certain degree of normal protein activity,This demonstrates the broad applicability of the method.
More compelling validation came from in vivo experiments. The research team tested PERT in a mouse model carrying a nonsense mutation in the IDUA gene, which leads to a severe disease analogous to human Hurler syndrome, characterized by the accumulation of toxic waste products within cells.
The results showed that PERT restored the production of full-length IDUA protein to only up to 7.6% of normal levels, a seemingly low level of restorationsufficient to significantly alleviate symptoms in miceSpecific manifestations included significant improvement in histopathological findings, reduced pathological accumulation of glycosaminoglycans (GAGs), and amelioration of pathological conditions in brain tissue and the liver; moreover, mice in the treatment group maintained normal body weight with no evident signs of toxicity.
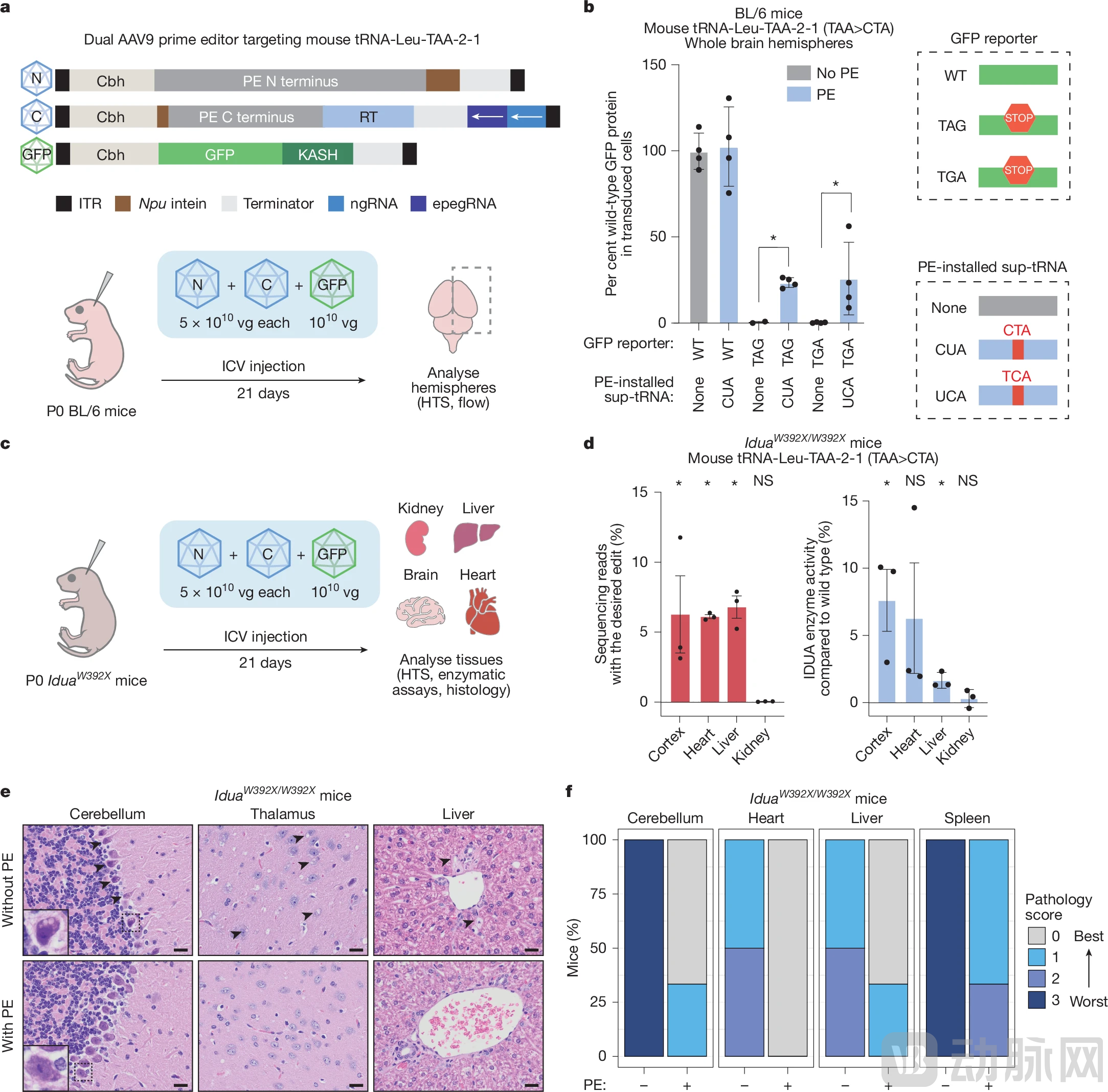
Figure: Prime editing generates functional sup-tRNA, exerting effects in animal disease models (Source: Nature)
This result indicates that for many genetic diseases, the tool can deliver clinical benefits without fully restoring normal protein levels, thereby significantly widening the therapeutic window of PERT.
For any gene-editing therapy, safety is the primary concern. The research team also conductedComprehensive Safety Assessment, through whole-proteome mass spectrometry analysis, researchers evaluated the 69 most abundant human proteins using TAG stop codons, and the results showed that sup-tRNA did not cause significant readthrough of natural stop codons.
Full transcriptome and proteome analyses in human cells and in the liver and cerebral cortex tissues of mice also revealed no significant changes, indicating that sup-tRNA expression does not substantially interfere with normal cellular functions. Off-target screening using lentiviral vectors further demonstrated that the Prime editor did not induce significant off-target editing.
These safety data have laid a solid foundation for the clinical translation of PERT.
The most significant breakthrough of PERT lies in its disease-agnostic nature. Traditional gene-editing therapies require the design of mutation-specific treatment regimens for each pathogenic mutation, meaning that even if two patients both have cystic fibrosis, they may require different treatments if their mutation sites differ.
In contrast, PERT overcomes this limitation: as long as the nonsense mutation is caused by the same type of stop codon (e.g., TAG),Regardless of the specific gene involved or the disease caused, it can in principle be treated with the same PERT tool.
Given that nonsense mutations account for 24% of known pathogenic mutations, PERT has the potential to treat hundreds of genetic diseases, covering a large patient population. This will significantly reduce drug development costs and time, enabling more patients with rare diseases to access therapeutic opportunities.
Moreover, unlike lipid nanoparticle approaches that require repeated administration,PERT achieves a one-time, permanent cure through gene editing.Once endogenous tRNA is successfully converted into sup-tRNA, the patient’s cells will continuously produce this therapeutic tRNA throughout their lifetime, eliminating the need for repeated treatments. This not only reduces the burden on patients but also significantly lowers long-term treatment costs.
David Liu paints a compelling vision of the future:“One day, medical centers could have a set of PERT medications ready in the refrigerator, available for immediate use. That is the dream.”In this vision, when a patient with a nonsense mutation-related disease presents to the hospital, physicians need only identify the specific type of premature stop codon (TAG, TAA, or TGA) to select the corresponding therapeutic regimen from a repository of PERT tools, thereby enabling rapid and standardized treatment.
As Zoya Ignatova, a biochemist at the University of Hamburg, commented, “It is encouraging to have another new technology. This will increase the chances of one of these methods entering clinical practice.” She is also involved in the development of suppressor tRNA therapy.
However, Ignatova also cautioned that suppressor tRNAs may be more complex than they initially appear. A given sup-tRNA is not effective against all nonsense mutations. Furthermore, optimal tRNA levels may vary across different tissues, meaning that a sup-tRNA therapy developed for treating lung conditions might not be safe or effective in the liver. “It won’t be a one-tRNA-fits-all solution,” she said.
David Liu also admitted that it will still take several years before PERT is truly used in patient treatment. “It will take years to make this a reality, but at least this work shows that it is biologically possible,” he said.
Overall, the emergence of PERT marks a new phase in therapeutic gene editing.From early gene knockout to precise editing, and now to disease-agnostic strategies, gene-editing technology is evolving from personalized customization toward modular universality.
As David Liu stated, “disease-agnostic approaches” represent an exciting new direction for therapeutic genome editing. Although there is still a long road from the laboratory to the clinic, PERT has already demonstrated biological feasibility. In the near future, when medical centers are truly able to stock a set of off-the-shelf PERT tools, patients with hundreds of genetic diseases will see the dawn of treatment.