Inocras Announces IPO Following Landmark Nature Publication on Whole-Genome Sequencing of 1,364 Breast Cancers
Breast cancer, as one of the most common malignant tumors in women worldwide, presents significant challenges to precision therapy due to its molecular heterogeneity and complex genomic features.
Recently, a research team composed of Samsung Medical Center, Seoul St. Mary's Hospital, and the bioinformatics company InocrasPublished in Nature, the largest whole-genome sequencing study of breast cancer to date.

(Source: Nature)
This study performed whole-genome sequencing (WGS) on 1,364 breast cancer patients, integrated with comprehensive clinical data, identified over 10.9 million somatic mutations, and revealed the predictive value of homologous recombination deficiency (HRD) across different treatment contexts, andRevealing That Tumor Genomic Instability Can Be Traced Back to Early Adolescence.
Professor Yeon Hee Park of Samsung Medical Center, the corresponding author of the study, stated, “Integrating genomic data with detailed clinical outcomes paves the way for more personalized and effective treatment strategies, with the ultimate goal of improving patient prognosis.”
Breast cancer can be classified into five molecular subtypes based on gene expression profiles: luminal A, luminal B, HER2-enriched, basal-like, and normal-like. There are significant differences in treatment strategies and prognosis among these subtypes. However, recurrence and metastasis remain major clinical challenges, highlighting the urgent need for a deeper understanding of the genomic characteristics of breast cancer.
Traditional genomic research has primarily relied on targeted sequencing technologies,Although such approaches focus on individual mutations in known oncogenes, they overlook a substantial amount of critical information beyond these targets. Pattern-driven genomic features, such as genomic rearrangements, copy number alterations (CNAs), and mutational signatures, are often not captured by traditional methods.
In contrast,Whole-genome sequencing as a more comprehensive technology, enabling the capture of the full spectrum of genomic alterations and providing an unbiased view of the cancer genome, thereby opening up new possibilities for biological discoveries and the exploration of potential biomarkers.
Although the academic community has analyzed a substantial number of cancer genomes over the past few decades, the clinical significance of these studies has often been limited by inadequate integration with clinical records. To truly unlock the practical value of genomic sequencing,The key lies in the organic integration of genomic data with comprehensive medical records, encompassing multiple dimensions such as treatment response, disease recurrence, and long-term clinical outcomes.Furthermore, previous genomic studies of breast cancer have been constrained by relatively limited sample sizes, which has hindered in-depth exploration of low-frequency mutations and subtype-specific variants.
It is worth noting that,There are significant differences in the molecular characteristics of breast cancer between Eastern and Western populations.Breast cancer patients in East Asian populations, such as those in South Korea, are generally younger at onset and have a lower proportion of estrogen receptor-positive (ER+) cases, which lends unique scientific value and clinical significance to large-scale genomic studies focused on Asian populations.
The CUBRICS cohort established in this study precisely fills this gap. As the largest breast cancer research cohort to date integrating whole-genome sequencing with comprehensive clinical data, it features a median patient age of only 44 years, significantly lower than that in Western countries, thereby providing an ideal platform for exploring the genomic characteristics of this unique population. The research team employed Inocras’s proprietary CancerVision platform to analyze all samples. This platform not only efficiently processes tumor-normal paired samples, ensuring high clinical accuracy and scalability, but also supports the deep integration of raw whole-genome sequencing data with meticulously curated clinical records, laying a solid foundation for subsequent analyses.
This study included patients from Samsung Medical Center and Seoul St. Mary's Hospital1,364 Breast Cancer Patients, recruited through prospective and retrospective cohorts between 2012 and 2023. Transcriptome sequencing was performed concurrently in 88.6% of cases (1,209 cases), enabling the research team to stratify cancers into five PAM50 subtypes and track the expression of acquired genomic variants.
Through whole-genome sequencing, the research team identified10929118 Somatic Mutations, including 8,935,132 single nucleotide variants (SNVs), 1,785,446 insertions and deletions (Indels), and 208,540 structural variants (SVs), with a median tumor mutational burden (TMB) of 4,742 mutations. The study applied the IntOGen pipeline to identify41 Breast Cancer Driver Genes, including four newly identified candidate genes, such as BCL11B, which was mutated in 23 patients at a frequency significantly higher than expected by chance.
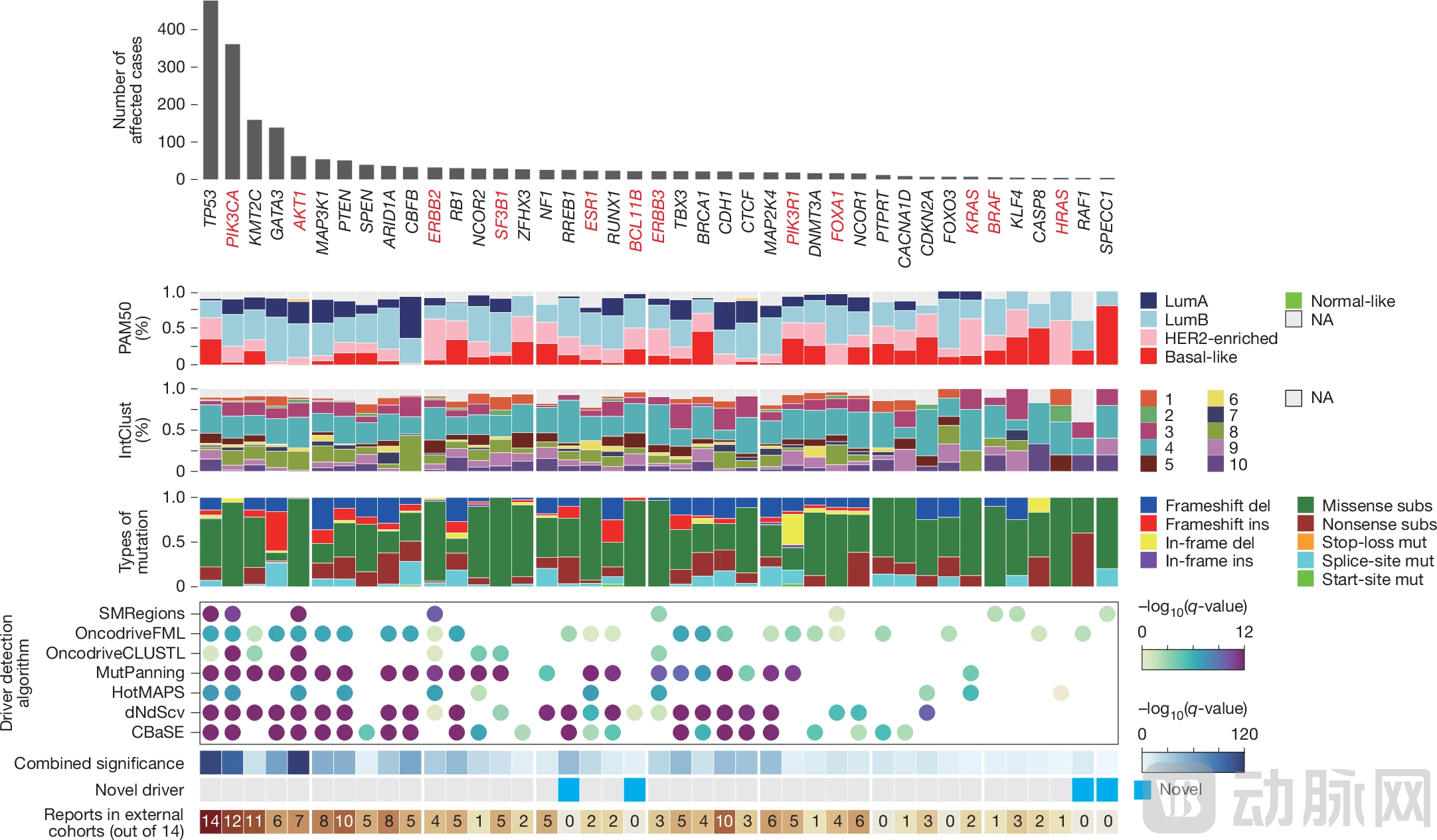
Figure: Driver genes in breast cancer (Source: Nature)
Mutational signature analysis identified 17 single-nucleotide variant signatures, 9 insertion/deletion signatures, and 6 structural variant signatures. Mutational signatures associated with homologous recombination deficiency (HRD) (SBS3, SBS8, ID6, SV3, and SV5) are particularly important, as they arePotential Predictive Biomarkers for Response to PARP Inhibitor Therapy.
HRD Exhibits Diametrically Opposed Prognostic Effects in Different Therapeutic Contexts.Among 89 patients with triple-negative breast cancer (TNBC) who received anthracycline-cyclophosphamide-based adjuvant chemotherapy, those with homologous recombination deficiency (HRD) (n=66) had significantly longer disease-free survival than those with proficient homologous recombination (HRP), with a hazard ratio of 0.10, confirming the high sensitivity of HRD tumors to DNA-damaging chemotherapy. However, among 57 patients with hormone receptor-positive advanced breast cancer treated with CDK4/6 inhibitors combined with endocrine therapy, 85% of the 13 HRD patients experienced disease progression, and their progression-free survival was significantly shorter than that of HRP patients, with a hazard ratio of 4.20. Multivariate Cox regression analysis showed that HRD was the most significant predictor of progression-free survival for this treatment regimen, with a hazard ratio of 10.20.
APOBEC-associated mutational signatures contributed to more than 10% of somatic single-nucleotide variants in 633 samples. The allele frequency of germline deletions in APOBEC3A and APOBEC3B was 31.8% (736 out of 1,364 cases) in this cohort, significantly higher than the 8.5% observed in European populations (P < 0.001).This variant is enriched in East Asian populations.Patients harboring this deletion exhibited higher TMB (median 5148 vs. 4325, P<0.001).
Structural variant analysis revealed that 15 breast cancer cases harbored translocations between chromosomes 8 and 11, placing the CCND1 gene in close proximity to the ZNF703/FGFR1 locus, which may promote oncogene expression through an enhancer hijacking mechanism. Recurrent fusions in luminal breast cancer included MIPOL1-TTC6 (9 cases), CEP112-PRKCA (6 cases), and CCDC170-ESR1 (6 cases), whereas basal-like breast cancer was predominantly characterized by BCL2L14-ETV6 (12 cases), AGO2-PTK2 (6 cases), and BRD4-NOTCH3 (6 cases). The CCDC170-ESR1 fusion has been confirmed to be associated with endocrine therapy resistance and metastasis.
Time analysis reveals,Most recurrent long-segment copy number amplification patterns are acquired at the emergence of the most recent common ancestor cell in cancer, decades before clinical diagnosis.The researchers pointed out: “This means that long-segment copy number amplification is an early evolutionary event in breast cancer, presumably occurring as early as early puberty.” This finding suggests that it may take decades from the initial genomic instability events to complete malignant transformation.
Furthermore, extrachromosomal DNA (ecDNA)-driven ERBB2 amplification demonstrates distinct predictive value. In the TransNEO cohort of 168 patients with HER2-positive breast cancer, none of the patients lacking focal ERBB2 amplification achieved a pathological complete response to neoadjuvant chemotherapy, whereas 3 out of 4 patients (75%) with focal amplification attained a complete response.
This study reveals the potential of whole-genome sequencing in advancing precision oncology for breast cancer, providing important insights for clinical decision-making.
HRD as a predictive biomarker exhibits treatment context dependency.In the adjuvant chemotherapy for triple-negative breast cancer, homologous recombination deficiency (HRD) predicts a better prognosis, consistent with the understanding that tumors with DNA damage repair defects are highly sensitive to chemotherapy; however, in the treatment of hormone receptor-positive advanced breast cancer with CDK4/6 inhibitors, HRD is associated with a poorer prognosis. This context-dependency underscores the importance of precisely evaluating biomarkers within specific clinical settings.
Quantitative Assessment of Tumor Heterogeneity Demonstrates Clinical Value.The Mutant Allele Tumor Heterogeneity (MATH) score is associated with overall survival, particularly in TP53-mutated tumors, where a high MATH score correlates with a poorer prognosis. Compared with traditional pathological assessment, whole-genome sequencing provides a more comprehensive characterization of tumor heterogeneity by capturing the genetic diversity arising from subclonal mutations.
By analyzing the timing of copy number variations, the research team reconstructed the temporal trajectory of cancer evolution.Revealing that genomic instability occurs decades before tumor diagnosis.This discovery expands genomic research from the traditional “two-dimensional plane” to a “three-dimensional spatiotemporal” framework, providing new perspectives for understanding cancer biology, the multistage evolutionary process of tumorigenesis, and the dynamic evolution of drug resistance mechanisms during treatment.
The discovery of racial differences is of great significance.The frequency of APOBEC3A/B deletion in the Korean population (31.8%) is significantly higher than that in the European population (8.5%)., and is associated with a higher tumor mutational burden. This suggests that individuals of different ethnicities may face distinct genomic risk profiles due to differences in genetic background, necessitating the development of differentiated risk assessment strategies and personalized treatment regimens.
The study demonstrates thatA New Paradigm: Whole-Genome Sequencing Combined with Real-World Clinical Data. By integrating large-scale whole-genome data with clinical records, data-driven biomarker discovery was achieved in retrospective analyses, reducing research costs and timelines, and providing a new pathway for accelerating the clinical translation of biomarkers.
Looking Ahead,Prospective clinical trials remain important for validating the functional significance of these genomic alterations.Quantitative assessment of tumor heterogeneity based on whole-genome sequencing will play a central role in shaping future precision oncology strategies. With declining sequencing costs, maturing analytical technologies, and the application of artificial intelligence in genomic data interpretation, the prospect of integrating whole-genome sequencing into routine cancer diagnosis and treatment is becoming increasingly clear. The resource established by this study, comprising whole-genome and clinical data from 1,364 breast cancer cases, will serve as a reference dataset for future intelligent cancer genomics platforms.
This largest-scale whole-genome study of breast cancer,By integrating over 10.9 million mutations with detailed clinical data, a comprehensive atlas of breast cancer genomic evolution has been mapped.From genomic instability events in early puberty to tumor diagnosis decades later, and from the dual role of homologous recombination deficiency (HRD) in different therapeutic contexts to extrachromosomal DNA-driven treatment responses, these findings have deepened our understanding of the molecular mechanisms of breast cancer and laid the foundation for the development of precision oncology. With the widespread adoption of whole-genome sequencing technologies and the integration of multi-omics data, personalized treatment based on patients’ molecular profiles will transition from vision to reality, ultimately improving prognoses for breast cancer patients.