Otsuka's Centanafadine: First-in-Class Non-Stimulant ADHD Therapy Receives FDA Priority Review
Recently, Otsuka Pharmaceutical announced that the U.S. FDA has accepted its New Drug Application (NDA) for the investigational drug centanafadine and granted it Priority Review designation. Centanafadine is a once-daily extended-release capsule and a potential first-in-class norepinephrine-dopamine-serotonin reuptake inhibitor (NDSRI), intended for the treatment of attention-deficit/hyperactivity disorder (ADHD) in children, adolescents, and adults.
The PDUFA date for this NDA is July 24, 2026. Once approved, centanafadine will become the first FDA-approved NDRI medication and the first new-mechanism, non-stimulant therapy for ADHD in over two decades.
In August 2025, Centanafadine sustained-release capsules received implicit approval for clinical trials from the Center for Drug Evaluation (CDE), with the intended indication being the treatment of attention-deficit/hyperactivity disorder (ADHD) in children and adolescents.
1ADHD Field: Global First-in-Class Triple Reuptake Inhibitor
ADHD is a chronic neurodevelopmental disorder common in school-aged children, primarily characterized by deficits in attention, hyperactivity, and impulsivity. Symptoms persist into adulthood in 30% to 50% of affected individuals. Since the release of the Diagnostic and Statistical Manual of Mental Disorders, Fifth Edition (DSM-5) in the United States in 2013, ADHD has been widely recognized as a condition that affects individuals across their entire lifespan. According to data from the U.S. Centers for Disease Control and Prevention (CDC), ADHD affects approximately 7 million children and an estimated 15.5 million adults in the United States.
Currently, the etiology and pathogenesis of ADHD remain incompletely understood. Among the leading biological hypotheses is the imbalance/dysfunction of monoamine neurotransmitters in the brain: In the mid-20th century, laboratory studies found that central nervous system stimulants could alleviate ADHD symptoms, which subsequently led to various hypotheses, including insufficient norepinephrine (NE) function, insufficient dopamine (DA) function, and either overactivation or relative deficiency of serotonin (5-HT) function.
Centanafadine is a potential “first-in-class” norepinephrine, dopamine, and serotonin reuptake inhibitor (NDSRI), with its core breakthrough lying in the pioneering simultaneous modulation of three neurotransmitter pathways.
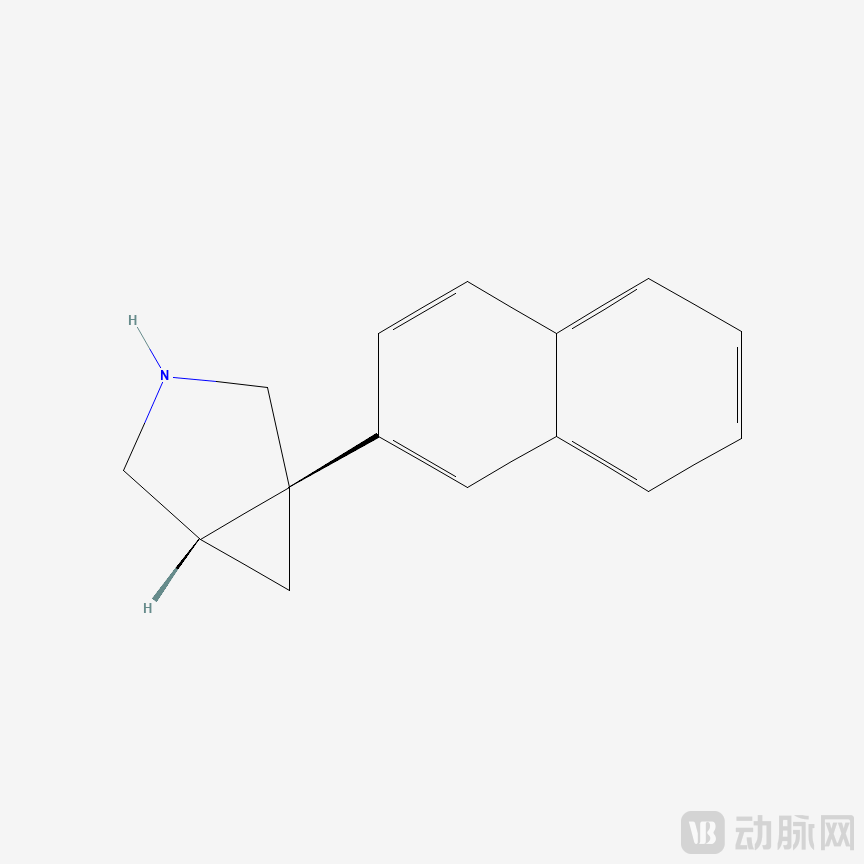
Chemical name: (1R,5S)-1-(naphthalen-2-yl)-3-azabicyclo[3.1.0]hexane; Molecular formula: C15H15N
Centanafadine is an unbalanced triple reuptake inhibitor with the highest potency for the norepinephrine transporter (NET), one-sixth the potency for the dopamine transporter (DAT), and one-fourteenth the potency for the serotonin transporter (SERT).
Specifically, Centanafadine is a new chemical entity classified as a BCS Class I molecule, characterized by high solubility and high permeability. Due to its relatively high therapeutic dose, the development of pellet-based sustained- and controlled-release formulations for Centanafadine presents significant challenges. Consequently, the R&D team has developed a novel pharmaceutical formulation: multiphase controlled-release pellets or granules with high drug loading or high payload capacity, designed to provide sustained therapeutic efficacy in disease treatment.
This also aligns with the demand for long-acting, sustained-release formulations in innovative ADHD medications. ADHD symptoms persist throughout the day, requiring continuous control from morning awakening through daytime learning, work, and social activities, to evening homework and daily routines. However, traditional sustained-release medications often cause symptom rebound due to a sharp decline in active ingredient concentrations in the evening. Meanwhile, significant fluctuations in plasma drug concentration can lead to unstable therapeutic efficacy and an increased risk of side effects.
As of February 2026, no norepinephrine-dopamine-serotonin triple reuptake inhibitor (NDSRI) has received final approval from the U.S. Food and Drug Administration (FDA).
In the same drug class, Luye Pharma’s Ansofaxine (desvenlafaxine hydrochloride extended-release tablets; brand name Ruoxinlin) is the first triple reuptake inhibitor approved in China (2022). As a serotonin-norepinephrine-dopamine reuptake inhibitor (SNDRI), it is a first-in-class product. Its approved indication is for the treatment of depression. On January 8, 2026, the Center for Drug Evaluation (CDE) website indicated that its marketing application for a new indication had been accepted; the anticipated indication is generalized anxiety disorder, which is currently in Phase III clinical trials. Currently, the U.S. marketing application for Ansofaxine remains under review.
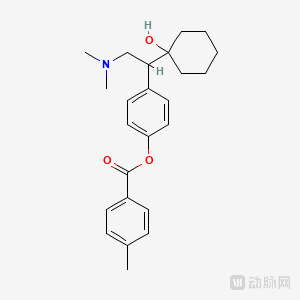
Chemical structure: (±)-4-[2-(dimethylamino)-1-(1-hydroxycyclohexyl)ethyl]phenyl 4-methylbenzoate hydrochloride dihydrate
From the perspective of its mechanism of action, Ansofaxine is a prodrug that is rapidly converted in vivo into its active metabolite, desvenlafaxine (a serotonin-norepinephrine reuptake inhibitor, SNRI). Ansofaxine itself possesses unique pharmacological characteristics; in addition to its affinity for serotonin and norepinephrine transporters, it also exhibits affinity for the dopamine transporter. Due to its lipophilicity, Ansofaxine and its metabolite desvenlafaxine can coexist in both blood and brain tissue, thereby achieving a combined therapeutic effect. The IC50 (half-maximal inhibitory concentration) refers to the drug concentration required to achieve 50% inhibition in in vitro experiments. One study, based on IC50 values, reported an inhibition ratio for SERT:NET:DAT of approximately 23:1.2:1.
Notably, the launch of Ansofaxine broke the drought of approved innovative drugs for depression since 2013. Upon approval of its second indication, it will become one of the first innovative therapies for anxiety disorders globally in nearly 15 years. In China, from 2022 to 2025, Ansofaxine has served over 80,000 patients, emerging as the fastest-growing new antidepressant in terms of sales in recent years, and was included in the National Reimbursement Drug List for the first time at the end of 2024.
2Statistically significant differences were observed as early as Day 7, enabling differentiated assessment of clinical efficacy in adult patients.
Clinical studies have demonstrated that centanafadine significantly alleviates the core symptoms of ADHD in children, adolescents, and adults.The submission of this New Drug Application (NDA) is primarily supported by data from four pivotal Phase 3 clinical trials, which evaluated efficacy and safety across different patient populations: one trial in children (ClinicalTrials.gov identifier: NCT05428033), one in adolescents (ClinicalTrials.gov identifier: NCT05257265), and two in adults (ClinicalTrials.gov identifiers: NCT03605680; NCT03605836). Adolescent and pediatric patients were assessed using the ADHD Rating Scale-5 (ADHD-RS-5), while adult patients were assessed using the ADHD Investigator Symptom Rating Scale (AISRS).
These trial data demonstrate that centanafadine achieved statistically significant and clinically meaningful efficacy in improving ADHD symptoms compared with placebo. Furthermore, centanafadine exhibited a favorable safety and tolerability profile across studies, with a low risk of abuse. The most common adverse events in children and adolescents included decreased appetite, nausea, rash, fatigue, abdominal pain, and somnolence, whereas the more common adverse events in adults were decreased appetite and headache.
Notably, in two adult studies, 446 and 430 patients aged 18 to 55 years with moderate-to-severe ADHD were randomly assigned to receive centanafadine at doses of 200 mg/day and 400 mg/day, respectively. The primary endpoint in both studies was the change from baseline in the ADHD Investigator Symptom Rating Scale (AISRS) score at Day 42 (with negative changes indicating improvement).
Study 1 results showed that both dose groups demonstrated significant improvement in the total AISRS score compared with the placebo group on Day 42 (200 mg/day: least-squares mean difference, -3.16 [95% confidence interval, -5.79 to -0.51]; P=0.019; 400 mg/day: -2.74 [95% confidence interval, -5.35 to -0.14]; P=0.039),Efficacy was observed as early as Day 28.. Compared with placebo, the Cohen’s d effect sizes for AISRS scores in the 200 mg/day and 400 mg/day dose groups were -0.28 and -0.24, respectively.
The results of Study 2 also demonstrated that both doses significantly improved the total AISRS score compared with placebo on Day 42 (200 mg/day: least squares mean difference, -4.01 [95% confidence interval, -6.55 to -1.46]; P=0.002; 400 mg/day: -4.42 [95% confidence interval, -7.02 to -1.82]; P<0.001).A statistically significant difference in AISRS scores was observed as early as Day 7.Compared with placebo, the Cohen’s d effect sizes for AISRS scores in the 200 mg/day and 400 mg/day dose groups were -0.37 and -0.40, respectively.
3ADHD: Acute Supply-Demand Imbalance, Rising Adult Demand, and the Rise of Domestic Innovative Drugs
According to Frost & Sullivan data, the global prevalence of ADHD in children is approximately 7.2%, while the prevalence among children and adolescents in China reaches 6.3%, with the rate in males being about twice that in females. Although the overall prevalence of ADHD is relatively high, only 10% of children with ADHD seek medical consultation.
Meanwhile, as public and social media attention to mental health continues to rise, adult ADHD is beginning to receive greater recognition.
According to the “Expert Consensus on the Diagnosis and Treatment of Adult Attention-Deficit/Hyperactivity Disorder in China (2023 Edition),” the global prevalence of adult ADHD ranges from 0.6% to 7.3%, with a rate of 5.2% in the United States and 1.8% in Shenzhen, China. Notably, compared with children and adolescents, adult ADHD is more difficult to identify, is associated with greater comorbidity, and causes broader impairment in individuals’ social functioning. A meta-analysis indicated that 45.76% of adults with ADHD have comorbid depressive disorders, and 34% of patients with treatment-resistant depression have comorbid ADHD.
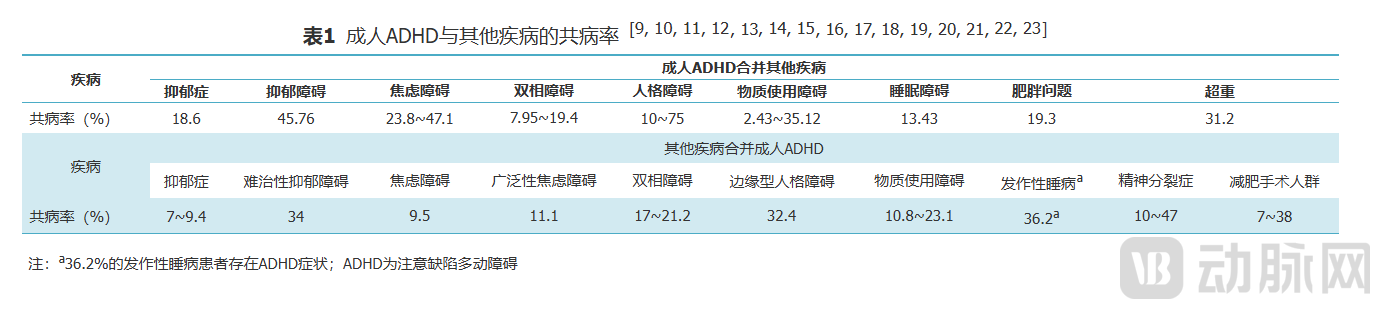
The Guidelines state that pharmacotherapy is one of the primary treatment modalities for adult ADHD. Currently, only long-acting methylphenidate extended-release formulations (central nervous system stimulants) are approved in China for the indication of adult ADHD and are recommended as first-line pharmacological treatment. Atomoxetine (a selective norepinephrine reuptake inhibitor) is approved in China solely for the treatment of ADHD in children and adolescents and serves as a second-line agent. When the aforementioned medications prove ineffective or are not tolerated, monotherapy or combination therapy with α2-adrenergic agonists (clonidine or guanfacine) is recommended as third-line treatment.
Overall, the current regulatory trend for ADHD medications is a gradual expansion from pediatric indications to the broader adolescent and adult markets.Johnson & Johnson/Janssen’s methylphenidate hydrochloride extended-release tablets (Concerta) were first launched in China in August 2005 for the treatment of pediatric ADHD; in April 2021, they received approval for an additional indication in adults and adolescents.
On the one hand, currently recommended medications, such as stimulants, carry risks of abuse or addiction, exhibit significant inter-individual variability in efficacy, and are associated with pronounced side effects (e.g., appetite suppression and insomnia). On the other hand, the heterogeneous clinical presentation of ADHD across patients underscores the importance of personalized treatment plans and various cutting-edge therapies.
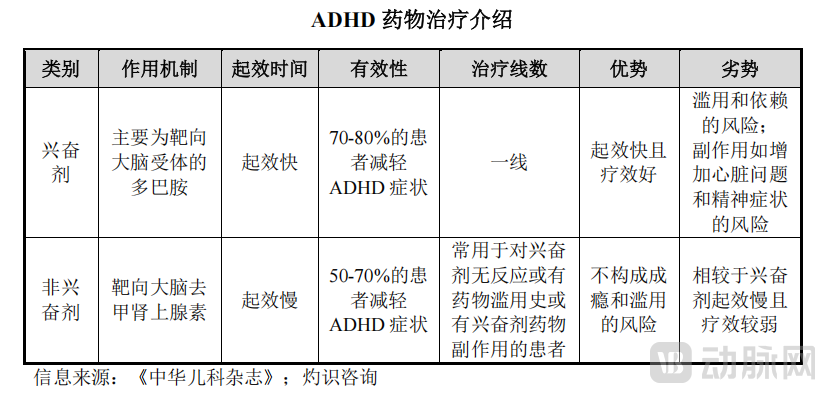
Source: Aikobai’s prospectus
From the perspective of drug supply dynamics, ADHD mirrors many other neurological disorders: early-market medications were predominantly introduced to China by multinational pharmaceutical companies in the early 21st century, maintaining long-term market monopoly. Currently, as patents for originator drugs expire sequentially and generic versions enter the market in large volumes, there is an increasingly urgent clinical demand for cutting-edge therapies and diversified medication regimens.Against this backdrop, if innovative drugs can achieve differentiation through formulation improvements (e.g., long-acting sustained-release), mechanistic breakthroughs (e.g., multi-pathway modulation), or targeting specific patient subgroups (e.g., adults or patients with comorbidities), they will rapidly penetrate the blue-ocean markets in specific scenarios upon approval, thereby establishing a competitive advantage through strategic positioning.
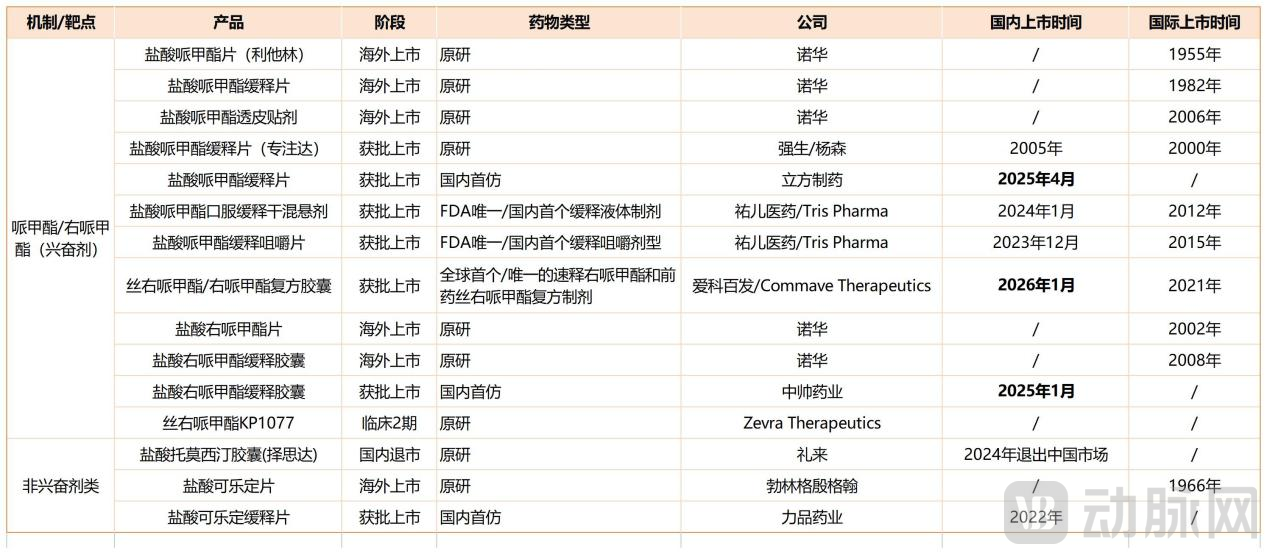 ADHD Drug Competitive Landscape (Graphic by VCBeat)
ADHD Drug Competitive Landscape (Graphic by VCBeat)
Note: There are numerous domestic generic versions of immediate-release methylphenidate tablets and atomoxetine hydrochloride capsules in China; these have been excluded from the statistics.
In January 2026, Aikobafa’s compound chlorpheniramine and dexmethylphenidate capsules (Aizhida) were approved in China for the treatment of patients aged 6 years and older with ADHD.As the world’s first and currently only combination formulation containing immediate-release dexmethylphenidate (d-MPH) and the prodrug serdexmethylphenidate (SDX), and as the first new-generation methylphenidate drug approved by the FDA in nearly two decades, it extends therapeutic efficacy to 13 hours through a dual mechanism.The drug was approved in the United States in March 2021, and in December of the same year, Aikobai Fa licensed it from the originator, Commave.
4About Otsuka Pharmaceutical
Otsuka Pharmaceutical was founded in 1921, starting as a small manufacturer of chemical raw materials. It began manufacturing and selling intravenous infusion products in 1946. In 1950, it spun off its organic chemistry division to establish Otsuka Chemical Co., Ltd. In 1963, it founded Taiho Pharmaceutical Co., Ltd., focusing on the oncology field. In 1964, Otsuka Pharmaceutical Co., Ltd. was established.
In the field of central nervous system (CNS) disorders, Otsuka Pharmaceutical launched ABILIFY Maintena in the United States in 2013, a once-monthly extended-release injectable antipsychotic that is currently marketed in more than 50 countries and regions. Its successor product, Rexulti, has been approved in the United States as an adjunctive treatment for major depressive disorder and schizophrenia and is sold in approximately 60 countries and regions. In 2023, Rexulti received FDA approval for the treatment of agitation associated with dementia due to Alzheimer’s disease, becoming the first and only drug indicated for this condition.