Roche's Hemlibra Gains EU Approval for Severe Hemophilia A Patients Without Factor VIII Inhibitors, Paving the Way for $4B Annual Sales
Mar 15, 2019 08:35
CST Updated
08:35

Roche
Oncology Drug Research, Development, and Manufacturing

European Commission
The European Commission, abbreviated as the EU Commission, is a supranational body under the European Union. Within the EU political system, the European Commission primarily undertakes executive tasks, thus being roughly equivalent to the government in a national system. However, the European Commission has other functions as well. In particular, except for the few circumstances specified in the treaties, the European Commission is the only institution with legislative power in the EU legislative process.

March 15, 2019 /BioValleyBIOON/ -- Swiss pharmaceutical giant Roche recently announced that the European Commission (EC) has approved Hemlibra (emicizumab) as a routine prophylactic treatment for adult and pediatric patients with severe hemophilia A (congenital factor VIII deficiency, FVIII <1%) who do not have factor VIII inhibitors, to prevent or reduce the frequency of bleeding episodes. Furthermore, the EC has approved multiple dosing regimens for Hemlibra (once weekly, once every two weeks, or once every four weeks, via subcutaneous injection) for all patients with hemophilia A eligible for this medication, including those who have developed factor VIII inhibitors.
In the United States and the European Union, Hemlibra was approved in November 2017 and February 2018, respectively, as a routine prophylactic treatment to reduce the frequency of bleeding episodes in adult and pediatric patients with hemophilia A who have developed inhibitors against coagulation factor VIII. In October 2018, Hemlibra received further approval from the U.S.FDAApproved as a routine prophylactic treatment to prevent or reduce the frequency of bleeding episodes in adult, pediatric, neonatal, and elderly patients with hemophilia A who do not have inhibitors to coagulation factor VIII. This approval makes Hemlibra the first new class of therapy approved in nearly 20 years for patients with severe hemophilia A without factor VIII inhibitors. It is also the only prophylactic treatment available for self-administered subcutaneous injection in patients with hemophilia A, regardless of inhibitor status, offering flexible dosing regimens (once weekly, once every two weeks, or once every four weeks via subcutaneous injection).
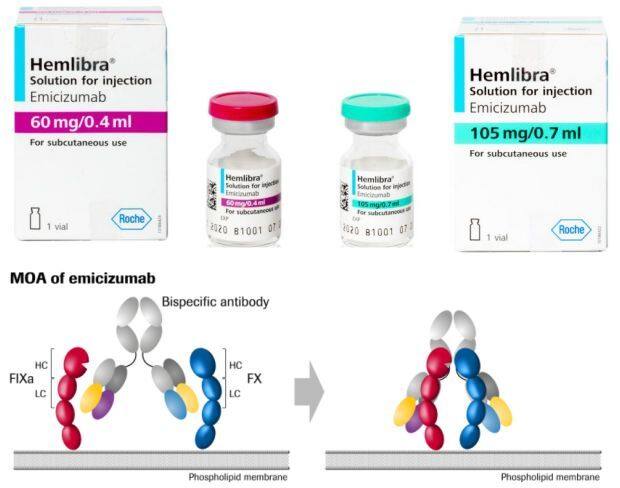
Hemlibra is a bispecific monoclonal antibody that brings together two proteins—coagulation factors IXa and X—that are essential for activating the natural coagulation cascade and restoring the natural coagulation process, thereby restoring hemostasis in patients with hemophilia A. Clinical studies have demonstrated that Hemlibra significantly reduces bleeding episodes and improves physical function.
Hemlibra was developed by Chugai Pharmaceutical and is currently being co-developed by Chugai Pharmaceutical, Roche, and its subsidiary Genentech. The development of this drug aims to help overcome the current clinical challenges faced by patients with hemophilia A: short duration of action of existing drugs, the development of factor VIII inhibitors, and the need for frequent intravenous infusions.
It is worth noting that, according to Roche’s 2018 annual performance report, Hemlibra achieved sales of CHF 224 million in its first full year on the market. With the expansion of the indicated patient population and market growth, commercial sales of Hemlibra are projected to increase substantially. Previously, Clarivate Analytics predicted that Hemlibra’s sales would reach $4 billion in 2022.
Hemlibra has been studied in one of the largest pivotal clinical research programs to date, comprising four pivotal Phase III HAVEN studies (HAVEN-1, -2, -3, and -4). The expanded indication approval in the European Union is based on data from two Phase III clinical studies (HAVEN-3 and HAVEN-4).
The HAVEN-3 study was a randomized, multicenter, open-label Phase III trial conducted in adolescents and adults aged 12 years and older with hemophilia A who did not have factor VIII inhibitors. Study data showed that, compared with patients not receiving prophylactic treatment, those receiving Hemlibra prophylaxis once weekly or once every two weeks experienced a 96% (p<0.0001) and 97% (p<0.0001) significant reduction in bleeding events, respectively. Importantly, in another patient cohort within the study, individuals who had previously received factor VIII prophylaxis in a non-interventional study switched to Hemlibra prophylaxis, enabling an intra-patient comparison (i.e., within the same patient) of the two prophylactic regimens. Based on this intra-patient comparison, Hemlibra significantly reduced bleeding events by 68% (p<0.0001), making it the first agent to demonstrate superior efficacy compared with the previous standard-of-care factor VIII prophylaxis. Regarding safety, no unexpected or serious adverse events related to Hemlibra were observed in the study, and common adverse events were consistent with those reported in previous studies.
The HAVEN-4 study is a single-arm, multicenter, open-label Phase III study evaluating the efficacy, safety, and pharmacokinetics (PK) of Hemlibra administered subcutaneously once every 4 weeks. The study enrolled 48 patients with hemophilia A (aged 12 years and older), either with or without factor VIII inhibitors, who had previously received on-demand or prophylactic treatment with factor VIII or bypassing agents. The study comprised two parts: a PK lead-in phase and an extension cohort. All patients entering the PK lead-in phase (n=7) had previously received on-demand treatment; these patients received a single 6 mg/kg subcutaneous dose of Hemlibra within the first 4 weeks, followed by 6 mg/kg every 4 weeks for at least 24 weeks to comprehensively characterize PK profiles. In the extension cohort (n=41), patients with factor VIII inhibitors (n=5) and those without factor VIII inhibitors (n=36) received 3 mg/kg weekly for 4 weeks, followed by 6 mg/kg every 4 weeks for at least 24 weeks. Per the study protocol, intermittent (episodic) treatment of breakthrough bleeding with factor VIII therapy or bypassing agents was permitted based on the patient’s factor VIII inhibitor status. Study results showed that among patients receiving Hemlibra prophylaxis once every 4 weeks, regardless of factor VIII inhibitor status, 56.1% experienced zero bleeds, and 90.2% experienced three or fewer treated bleeding events. (Bioon.com)