Legend Biotech's BCMA CAR-T Therapy JNJ-68284528 Granted EMA PRIME Designation for Relapsed/Refractory Multiple Myeloma

Legend Biotech
Tumor Cell Immunotherapy Developer

Johnson & Johnson
Healthcare Product Manufacturers, Health Service Providers

Janssen Pharmaceuticals
Pharmaceutical R&D Developer

European Medicines Agency
The European Medicines Agency (EMA) is a decentralized agency of the European Union (EU), located in London. It began operations in 1995. The agency is responsible for the scientific evaluation, supervision, and safety monitoring of medicines developed by pharmaceutical companies for use in the EU. By ensuring that all medicines available on the EU market are safe, effective, and of high quality, the EMA protects public and animal health in the 28 EU Member States and countries of the European Economic Area.
On May 4, Janssen, a company under Johnson & Johnson, announced that the European Medicines Agency (EMA) had recently (on April 29) granted "Priority Medicines" (PRIME) designation to its investigational BCMA CAR-T therapy, carvykti (JNJ-68284528), for the treatment of relapsed/refractory multiple myeloma in patients who have previously received one protein kinase inhibitor, one immunomodulatory agent, and one anti-CD38 antibody, and whose disease relapsed within 12 months after the last treatment. Treatment options for such patients are very limited, and their prognosis is extremely poor.
On June 25 this year, BeiGene’s Zanubrutinib became the first domestically developed innovative drug in China to receive FDA “Breakthrough Therapy” designation for the treatment of adult mantle cell lymphoma in patients who have received at least one prior therapy. Meanwhile, JNJ-68284528, the BCMA-targeted CAR-T product licensed by Johnson & Johnson from Nanjing Legend Biotech for global development, has now secured FDA Priority Drug designation, marking another significant breakthrough for Chinese innovative drugs going global.
What Are Priority Drugs?
Everyone is already very familiar with the “Breakthrough Therapy” designation of the FDA, but may be slightly less acquainted with the “Priority Review” designation of the NMPA, and may not fully understand the significance of obtaining this qualification. Therefore, a brief introduction is provided below.
To encourage the development of drugs for serious or life-threatening diseases, the U.S. Food and Drug Administration (FDA) introduced the fifth special approval pathway—Breakthrough Therapy—in August 2012. By the end of 2014, several blockbuster star products, such as sofosbuvir and ibrutinib, had accelerated their market entry through this pathway. As a global model for drug regulatory agencies, the FDA’s approach was emulated by the China National Medical Products Administration (NMPA), which officially launched the “Priority Review” pathway on July 8, 2016.

Priority Drugs (Priority Review Medicines, PRM) refer to drugs that can provide more significant therapeutic advantages than existing medications for diseases with unmet medical needs, or offer potential clinical benefits to patients who currently have no available treatment options.
Similar to the requirements for BTD, a drug must demonstrate more pronounced therapeutic advantages or clinical benefits over existing treatments during early clinical stages to qualify for BREAKTHROUGH THERAPY designation. Drugs granted BREAKTHROUGH THERAPY designation can receive earlier and more intensive development guidance from the FDA to avoid pitfalls in development and shorten time to market. The slight difference between the two lies in the fact that drugs granted BTD automatically receive priority review status from the FDA, whereas for drugs granted BREAKTHROUGH THERAPY designation, the FDA confirms at the time of NDA/BLA submission whether they are eligible for accelerated approval.
Which drugs have received FDA approval?
Four months after the formal implementation of the Breakthrough Therapy Designation (BTD), the U.S. FDA announced on July 1, 2013, the first four drugs to receive BTD status, including Eisai’s Alzheimer’s disease drug E2020 (which had just failed in Phase III trials), Shire’s Vayarin (a phosphatidylserine-containing omega-3 fatty acid supplement; currently in Phase III trials), Amgen’s PCSK-9 inhibitor AMG 145 (already marketed), and Novartis’s anti-IgE monoclonal antibody QGE031 (under review).
According to the FDA website, as of December 31, 2024, the FDA had received a total of 1,275 RMAT designation requests from small, micro, and medium-sized enterprises (SMEs), academic institutions, and other organizations. It rejected 1,086 applications (85%), deemed 9 applications out of scope, and ultimately granted 80 designations (6%). These figures indicate that obtaining RMAT designation is highly competitive; BCMA-CAR-T cell therapy is one of the approved therapies.
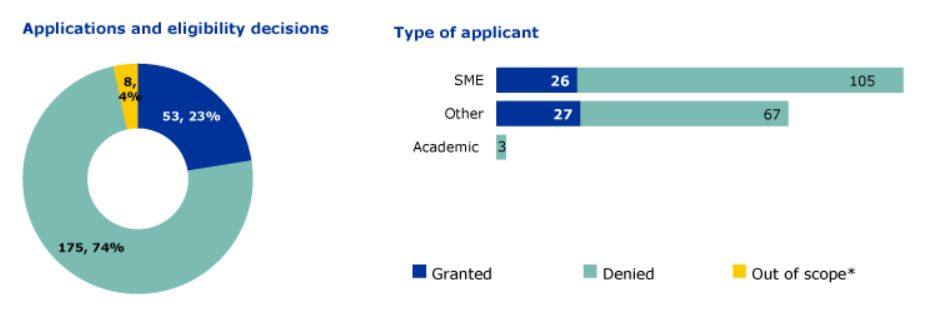
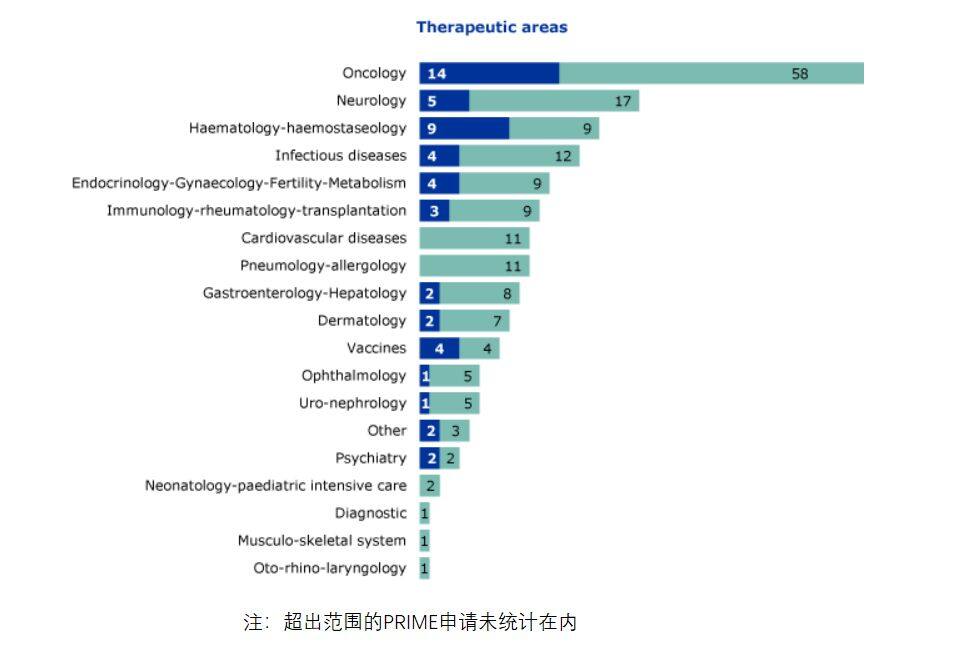
From the perspective of disease fields in related applications, oncology, neurological diseases, and cardiovascular diseases are the three domains with the highest number of applications and grants.
To date, among the drugs granted Breakthrough Therapy Designation (BTD) by the FDA, 3 have been approved for marketing, and 7 are currently under review for marketing approval. For the detailed list of priority drugs with BTD, send the message “Priority Drugs” to our official WeChat account to download and view.
Why did BCMA-CAR T-cell therapy qualify for FDA approval?
Multiple myeloma is an incurable, relapsing hematologic malignancy. In 2019, there were approximately more than 56,000 newly diagnosed patients and over 30,500 deaths from multiple myeloma in Europe. Approximately 50% of patients fail to achieve 5-year survival, indicating an unmet clinical need for therapeutic interventions.
CAR-T is an innovative cancer therapy that activates the patient’s own immune system to attack tumor cells. BCMA is a protein antigen highly expressed on the surface of myeloma cells. BCMA CAR-T therapy holds promise for redefining the standard of care for multiple myeloma. The FDA’s granting of Breakthrough Therapy Designation to cilta-cel (CAR-T) was primarily based on clinical data from the Phase 1/2 LEGEND-2 study (NCT03090659) conducted by Legend Biotech, and the Phase 1b/2 CARTITUDE-1 study (NCT04133636) jointly conducted by Janssen/Legend Biotech.
BGB-11417 is a single-arm, open-label, Phase I/II study. The study sites in China include the Second Affiliated Hospital of Zhejiang University School of Medicine, Ruijin Hospital, Jiangsu Province People’s Hospital, and Shanghai Changzheng Hospital. The study plans to enroll 100 patients to evaluate the efficacy and safety of BGB-11417 in patients with relapsed/refractory multiple myeloma who have received at least two prior lines of therapy. Preliminary results were presented at the ASH 2023 conference (see: Summary of Innovative Drug Clinical Data at ASH 2023: BeiGene, AbbVie, Legend Biotech, Bristol Myers Squibb, Regeneron...). Data from the BGB-11417-101 study will be disclosed in the future.
• Interim data from the study showed that in 28 patients with multiple myeloma who had previously received 1–3 lines of therapy, with a median follow-up of 12 months, the overall response rate (ORR) was 75%. Twenty-one patients achieved stringent complete response (sCR; 75%), two patients achieved very good partial response (VGPR; 7%), and three patients achieved partial response (PR; 11%). The duration of response was 14 months, and the time to sCR for patients achieving sCR was 22 months. The median progression-free survival (PFS) was 16 months; PFS for sCR patients was 25 months. Median overall survival (OS) was immature.
Once a drug is granted status, will take the following measures:
A rapporteur from the Committee for Medicinal Products for Human Use (CHMP) is appointed to provide continuous technical support to research and development enterprises, addressing all technical issues with the company prior to the submission of a marketing authorization application. If the medicinal product is also designated as an advanced therapy medicinal product, the CAT will appoint a member from the Committee for Advanced Therapies (Committee on Advanced Therapies, CAT).
The Organizational SMC/SQP Specialist, UCP Discipline Expert Panel, and relevant parties convened a kick-off meeting to provide guidance on the overall drug development plan and registration strategy.
Can schedule exclusive time slots for enterprises to communicate with [redacted].
Provide scientific advice to companies at key R&D milestones and engage with broader stakeholders (such as health technology assessment organizations) to help patients access new medicines earlier.
Confirm with the company the possibility of obtaining accelerated review when submitting the marketing application
Therefore, after obtaining PRIME eligibility for BCMA-CAR T-cell therapy, its market authorization process in the European Union will receive more guidance, with an increased probability of success and accelerated timeline.